


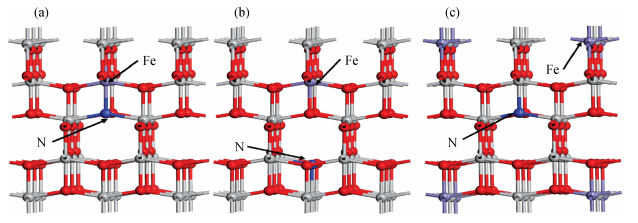
Fig. 1.
(Color online) Supercell models of N/Fe-codoped TiO$_{2}$. (a) N/Fe-1. (b) N/Fe-2. (c) N/Fe-3. The ion doping sites are marked with Fe and N. The gray and red spheres represent the Ti and O atoms,respectively.
SEMICONDUCTOR PHYSICS
Zhuomao Zhu1, Baoan Bian1 and Haifeng Shi1, 2
Abstract: The electronic structure and optical properties of N and Fe codoping TiO2 have been investigated by first-principles calculations based on density functional theory.The calculated results indicate that the stability of N and Fe codoping TiO2 will change at different substitutional sites of N and Fe.The mechanism of band gap narrowing of doping TiO2 is discussed by investigating the density of state.The different substitutional site of N and Fe in codoping TiO2 influences the visible-light absorption.An increased visible-light absorption for doping TiO2 results from the synergistic effect of N and Fe codoping.Therefore, N and Fe codoping may enhance the visible-light photocatalytic activity of TiO2.
Keywords: codoping, first-principle, electronic structure, optical properties
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] |

Table 1. Defect formation energy of different doping models.
| Doped system | Defect formation energy (eV)} | |
| O-rich | Ti-rich | |
| N-doped TiO$_{2}$ | 5.3353 | -0.4540 |
| Fe-doped TiO$_{2}$ | 0.8321 | 12.4109 |
| N/Fe-1 | 5.2868 | 11.0762 |
| N/Fe-2 | 5.7506 | 11.5400 |
| N/Fe-3 | 5.7516 | 11.5410 |
 DownLoad: CSV
DownLoad: CSV
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] |









Article views: 3537 Times PDF downloads: 26 Times Cited by: 0 Times
Received: 22 December 2014 Revised: Online: Published: 01 October 2015
| Citation: |
Zhuomao Zhu, Baoan Bian, Haifeng Shi. Effect of N and Fe codoping on the electronic structure and optical properties of TiO2 from first-principles study[J]. Journal of Semiconductors, 2015, 36(10): 102003. doi: 10.1088/1674-4926/36/10/102003
****
Z M Zhu, B A Bian, H F Shi. Effect of N and Fe codoping on the electronic structure and optical properties of TiO2 from first-principles study[J]. J. Semicond., 2015, 36(10): 102003. doi: 10.1088/1674-4926/36/10/102003.

|
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] |

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2


