


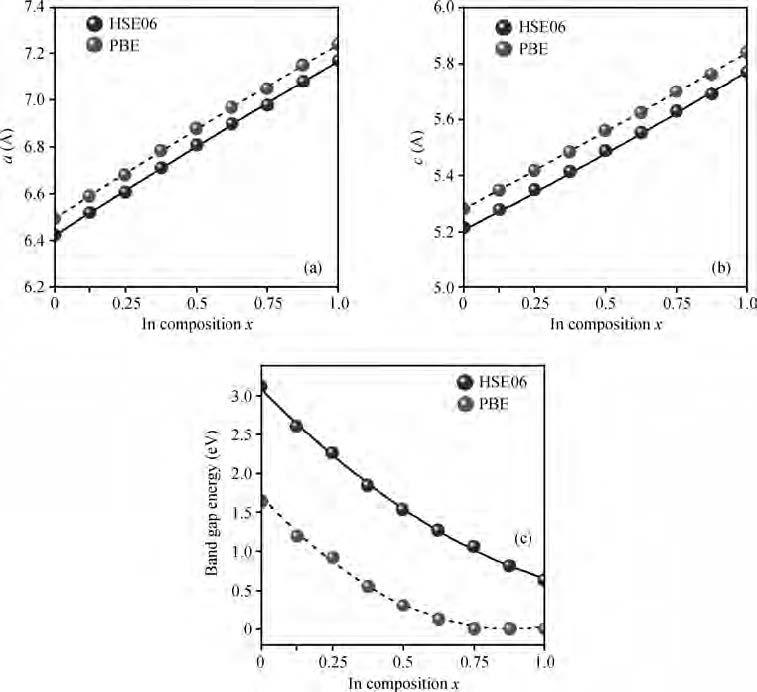
Fig. 1.
(a) Lattice constants $a$ as a function of the In composition $x.$ (b) Lattice constants $c$ as a function of the In content $x$. (c) Band gap as a function of the In composition $x$.
SEMICONDUCTOR PHYSICS
Mei Lin1, Yixu Xu1, Jianhua Zhang2, Shunqing Wu1 and Zizhong Zhu1, 3,
Corresponding author: Zizhong Zhu, Email:zzhu@xmu.edu.cn
Abstract: The electronic band structures and band gap bowing parameters of InxGa1-xN are studied by the first-principles method based on the density functional theory. Calculations by employing both the Heyd-Scuseria-Ernzerh of hybrid functional (HSE06) and the Perdew-Burke-Ernzerhof (PBE) one are performed. We found that the theoretical band gap bowing parameter is dependent significantly on the calculation method, especially on the exchange-correlation functional employed in the DFT calculations. The band gap of InxGa1-xN alloy decreases considerably when the In constituent x increases. It is the interactions of s-s and p-p orbitals between anions and cations that play significant roles in formatting the band gaps bowing. In general, the HSE06 hybrid functional could provide a good alternative to the PBE functional in calculating the band gap bowing parameters.
Keywords: InxGa1-xN, bowing parameters, HSE06 functional, PBE functional
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] |
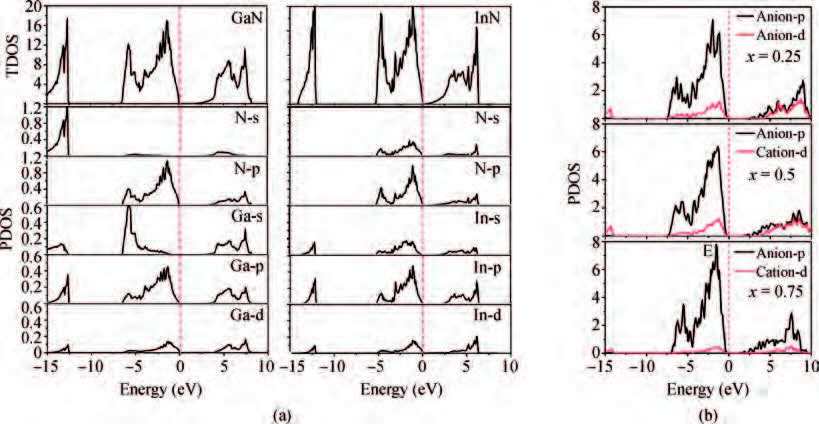
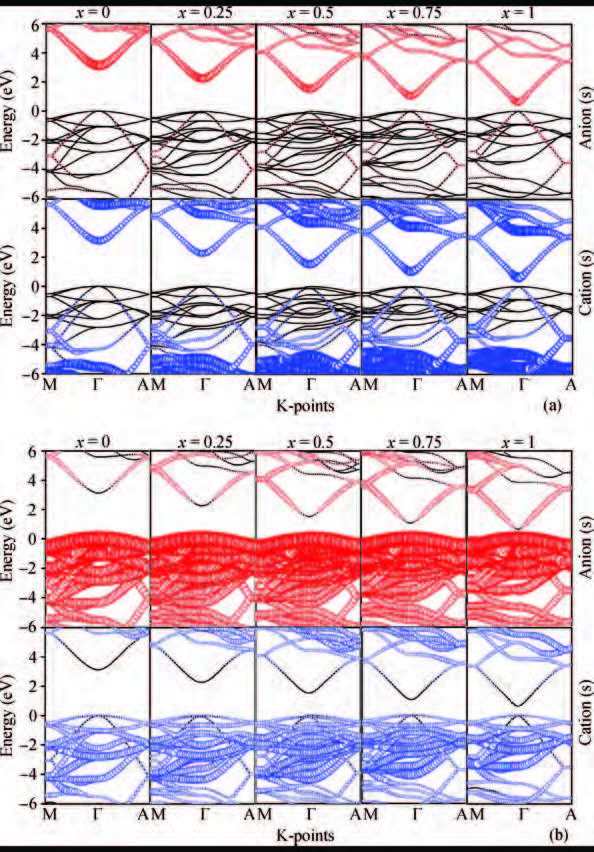
Table 1. Energy gaps and bowing parameters of In$_{x}$Ga$_{1-x}$N system calculated by PBE and HSE06 functional methods,compared with available theoretical and experimental data.
 DownLoad: CSV
DownLoad: CSV
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] |









Article views: 3053 Times PDF downloads: 24 Times Cited by: 0 Times
Received: 04 August 2015 Revised: Online: Published: 01 April 2016
| Citation: |
Mei Lin, Yixu Xu, Jianhua Zhang, Shunqing Wu, Zizhong Zhu. Hybrid functional calculations on the band gap bowing parameters of InxGa1-xN[J]. Journal of Semiconductors, 2016, 37(4): 042001. doi: 10.1088/1674-4926/37/4/042001
****
M Lin, Y X Xu, J H Zhang, S Q Wu, Z Z Zhu. Hybrid functional calculations on the band gap bowing parameters of InxGa1-xN[J]. J. Semicond., 2016, 37(4): 042001. doi: 10.1088/1674-4926/37/4/042001.

|
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] |

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2


