


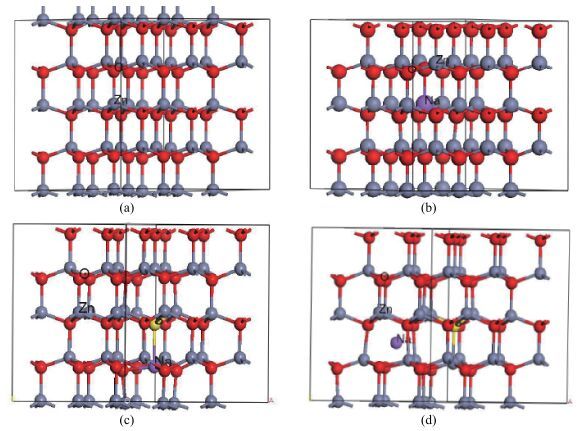
Fig. 1.
(Color online) Supercell of ZnO for (a) ZnO, (b) Na$_{\mathrm{Zn}}$ , (c) Na$_{\mathrm{Zn}}$ -S$_{\mathrm{O}}$ , and (d) Na$_{\mathrm{in}}$ -S$_{\mathrm{O}}$ .
SEMICONDUCTOR MATERIALS
Xingyi Tan, Qiang Li and Yongdan Zhu
Corresponding author: Xingyi Tan, Email:tanxingyi1009@163.com
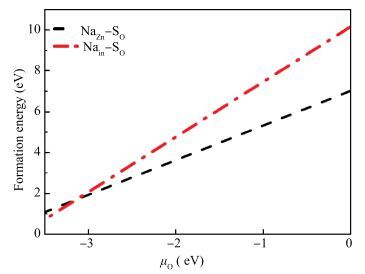
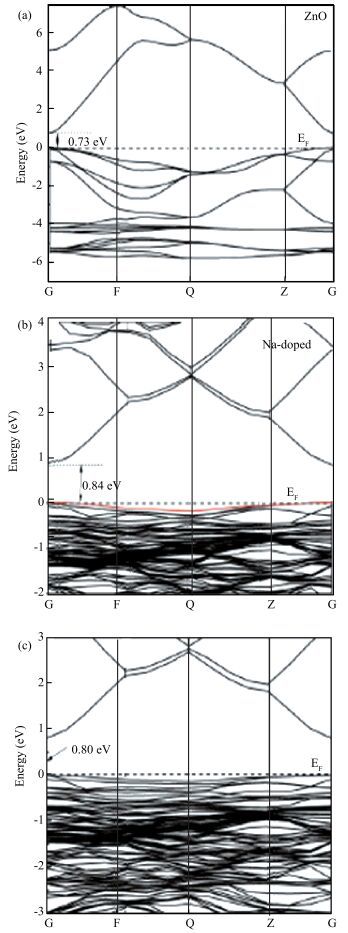
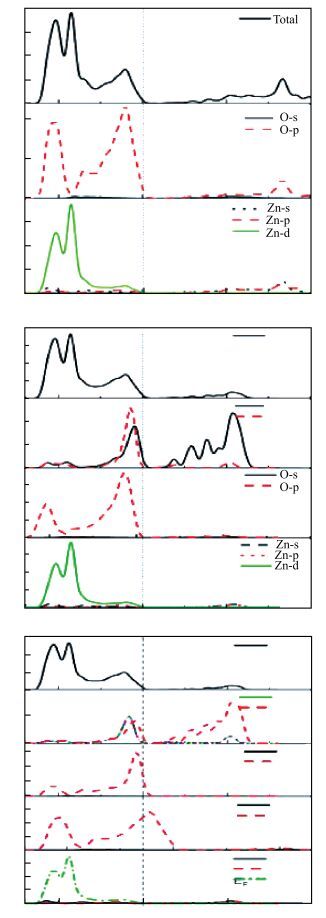
Abstract: Using the first-principles method based on the density functional theory, the formation energy, electronic structures of S-Na co-doping in ZnO were calculated. The calculated results show that NaZn-SO have smaller formation energy than Nain-SO in energy ranges from -3.10 to 0 eV of μO, indicating that it opens up a new opportunity for growth the p-type ZnO. The band structure shows that the NaZn system is a p-type direct-band-gap semiconductor material and the calculated band gap (0.84 eV) is larger than pure ZnO (0.74 eV). The NaZn-SO system is also a p-type semiconductor material with a direct band gap (0.80 eV). The influence of S-Na co-doping in ZnO on p-type conductivity is also discussed. The effective masses of NaZn-SO are larger than effective masses of NaZn and the NaZn-SO have more hole carriers than NaZn, meaning the hole in the NaZn-SO system may have a better carrier transfer character. So we inferred that NaZn-SO should be a candidate of p-type conduction.
Key words: p-type ZnO, electronic structures, conductive property
| [1] |
Özgür Ü, Alivov Y I, Liu C, et al. A comprehensive review of ZnO materials and devices. J Appl Phys, 2005, 98:0413011 https://www.researchgate.net/publication/276379347_A_Comprehensive_Review_of_ZnO_Materials_and_Devices
|
| [2] |
Sara K G, Ramin Y, Farid J S, et al. Optical and electrical properties of p-type Ag-doped ZnO nanostructures. Ceram Int, 2014, 40:7957 doi: 10.1016/j.ceramint.2013.12.145
|
| [3] |
Li W J, Kong C Y, Qin G P, et al. p-type conductivity and stability of Ag-N codoped ZnO thin films. J. Alloys Compd, 2014, 609:173 doi: 10.1016/j.jallcom.2014.04.051
|
| [4] |
Alivov Y I, Kalinina E V, Cherenkov A E, et al. Fabrication and characterization of n-ZnO/p-AlGaN heterojunction light-emitting diodes on 6H-SiC substrates. Appl Phys Lett, 2003, 83:4719 doi: 10.1063/1.1632537
|
| [5] |
Huang Y B, Zhou W, Wu P. Room-temperature ferromagnetism in epitaxial p-type K-doped ZnO films. Solid State Commun, 2014, 183:31 doi: 10.1016/j.ssc.2013.12.024
|
| [6] |
Makino T, Chia C H, Tuan N T, et al. Radiative and nonradiative recombination processes in lattice-matched (Cd, Zn)O/(Mg, Zn)O multiquantum wells. Appl Phys Lett, 2000, 77:1632 doi: 10.1063/1.1308540
|
| [7] |
Sáaedi A, Yousefi R, Jamali-Sheini F, et al. Optical and electrical properties of p-type Li-doped ZnO nanowires. Superlattices Microstruct, 2013, 61:91 doi: 10.1016/j.spmi.2013.06.014
|
| [8] |
Zhang S B, Wei S H, Zunger A. Intrinsic n-type versus p-type doping asymmetry and the defect physics of ZnO. Phys Rev B, 2001, 63:075205 doi: 10.1103/PhysRevB.63.075205
|
| [9] |
Dhara S, Giri P K. Stable p-type conductivity and enhanced photoconductivity from nitrogen-doped annealed ZnO thin film. Thin Solid Films, 2012, 520:5000 doi: 10.1016/j.tsf.2012.02.081
|
| [10] |
Liu J S, Shan C X, Shen H, et al. ZnO light-emitting devices with a lifetime of 6.8 hours. Appl Phys Lett, 2012, 101:011106 doi: 10.1063/1.4733298
|
| [11] |
Shen H, Shan C X, Liu J S, et al. Stable p-type ZnO films obtained by lithium-nitrogen codoping method. Phys Status Solidi B, 2013, 250:2102 https://www.researchgate.net/publication/264251371_Stable_p-type_ZnO_films_obtained_by_lithium-nitrogen_codoping_method
|
| [12] |
Park C H, Zhang S B, Wei S H. Origin of p-type doping difficulty in ZnO:the impurity perspective. Phys Rev B, 2002, 66:073202 doi: 10.1103/PhysRevB.66.073202
|
| [13] |
Yang X P, Lu J G, Zhang H H, et al. Preparation and XRD analyses of Na-doped ZnO nanorod arrays based on experiment and theory. Chem Phys Lett, 2012, 528(3):16 https://www.researchgate.net/publication/256683125_Preparation_and_XRD_Analyses_of_Na-Doped_ZnO_Nanorod_Arrays_Based_on_Experiment_and_Theory
|
| [14] |
Lin S S, Lu J G, Ye Z Z, et al. p-type behavior in Na-doped ZnO films and ZnO homojunction light-emitting diodes. Solid State Commun, 2008, 148:25 doi: 10.1016/j.ssc.2008.07.028
|
| [15] |
Pandey S K, Awasthi V, Sengar B S, et al. Band alignment and photon extraction studies of Na-doped MgZnO/Ga-doped ZnO heterojunction for light-emitter applications. J Appl Phys, 2015, 118:165301 doi: 10.1063/1.4934560
|
| [16] |
Persson C, Platzer-Björkman C, Malmström J, et al. Strong valence-band offset bowing of ZnO1-xSx enhances p-type nitrogen doping of ZnO-like alloys. Phys Rev Lett, 2006, 97:146403 doi: 10.1103/PhysRevLett.97.146403
|
| [17] |
Xu T N, Li X, Lu Z, et al. Realization of Ag-S codoped p-type ZnO thin films. Appl Sur Sci, 2014, 316:62 doi: 10.1016/j.apsusc.2014.07.156
|
| [18] |
Xu Y, Yang T, Yao B, et al. Influence of Ag-S codoping on silver chemical states and stable p-type conduction behavior of the ZnO films. Ceram Int, 2014, 40:2161 doi: 10.1016/j.ceramint.2013.07.133
|
| [19] |
Niu W Z, Xu H B, Guo Y M, et al. The effect of sulfur on the electrical properties of S and N co-doped ZnO thin films:experiment and first-principles calculations. Phys Chem Chem Phys, 2015, 17:16705 doi: 10.1039/C5CP02434J
|
| [20] |
Perdew J P, Burke S, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett, 1996, 77:3865 doi: 10.1103/PhysRevLett.77.3865
|
| [21] |
Van de Walle C G, Neugebauer J. First-principles calculations for defects and impurities:applications to Ⅲ-nitrides. J Appl Phys, 2004, 95:3851 doi: 10.1063/1.1682673
|
| [22] |
Oba F, Togo A, Tanaka I, et al. Defect energetics in ZnO:a hybrid Hartree-Fock density functional study. Phys Rev B, 2008, 77:245202 doi: 10.1103/PhysRevB.77.245202
|
| [23] |
Yan Y F, Zhang S B, Pantelides S T. Control of doping by impurity chemical potentials:predictions for p-type ZnO. Phys Rev Lett, 2001, 86:5723 doi: 10.1103/PhysRevLett.86.5723
|
| [24] |
Guo T T, Dong G B, Chena Q, et al. First-principles calculation on p-type conduction of (Sb, N) codoping in ZnO. J Phys Chem Solid, 2014, 75:42 doi: 10.1016/j.jpcs.2013.08.006
|
| [25] |
Zhang J K, Deng S H, Jin H, et al. First-principle study on the electronic structure and p-type conductivity of ZnO. Acta Phys Sin, 2007, 56:5371 https://www.researchgate.net/publication/291645152_First-principle_study_on_the_electronic_structure_and_p-type_conductivity_of_ZnO
|
| [26] |
Limpijumnong S, Zhang S B, Wei S H, et al. Doping by large-size-mismatched impurities:the microscopic origin of arsenic-or antimony-doped p-type zinc oxide. Phys Rev Lett, 2004, 92:155504 doi: 10.1103/PhysRevLett.92.155504
|
| [1] |
Özgür Ü, Alivov Y I, Liu C, et al. A comprehensive review of ZnO materials and devices. J Appl Phys, 2005, 98:0413011 https://www.researchgate.net/publication/276379347_A_Comprehensive_Review_of_ZnO_Materials_and_Devices
|
| [2] |
Sara K G, Ramin Y, Farid J S, et al. Optical and electrical properties of p-type Ag-doped ZnO nanostructures. Ceram Int, 2014, 40:7957 doi: 10.1016/j.ceramint.2013.12.145
|
| [3] |
Li W J, Kong C Y, Qin G P, et al. p-type conductivity and stability of Ag-N codoped ZnO thin films. J. Alloys Compd, 2014, 609:173 doi: 10.1016/j.jallcom.2014.04.051
|
| [4] |
Alivov Y I, Kalinina E V, Cherenkov A E, et al. Fabrication and characterization of n-ZnO/p-AlGaN heterojunction light-emitting diodes on 6H-SiC substrates. Appl Phys Lett, 2003, 83:4719 doi: 10.1063/1.1632537
|
| [5] |
Huang Y B, Zhou W, Wu P. Room-temperature ferromagnetism in epitaxial p-type K-doped ZnO films. Solid State Commun, 2014, 183:31 doi: 10.1016/j.ssc.2013.12.024
|
| [6] |
Makino T, Chia C H, Tuan N T, et al. Radiative and nonradiative recombination processes in lattice-matched (Cd, Zn)O/(Mg, Zn)O multiquantum wells. Appl Phys Lett, 2000, 77:1632 doi: 10.1063/1.1308540
|
| [7] |
Sáaedi A, Yousefi R, Jamali-Sheini F, et al. Optical and electrical properties of p-type Li-doped ZnO nanowires. Superlattices Microstruct, 2013, 61:91 doi: 10.1016/j.spmi.2013.06.014
|
| [8] |
Zhang S B, Wei S H, Zunger A. Intrinsic n-type versus p-type doping asymmetry and the defect physics of ZnO. Phys Rev B, 2001, 63:075205 doi: 10.1103/PhysRevB.63.075205
|
| [9] |
Dhara S, Giri P K. Stable p-type conductivity and enhanced photoconductivity from nitrogen-doped annealed ZnO thin film. Thin Solid Films, 2012, 520:5000 doi: 10.1016/j.tsf.2012.02.081
|
| [10] |
Liu J S, Shan C X, Shen H, et al. ZnO light-emitting devices with a lifetime of 6.8 hours. Appl Phys Lett, 2012, 101:011106 doi: 10.1063/1.4733298
|
| [11] |
Shen H, Shan C X, Liu J S, et al. Stable p-type ZnO films obtained by lithium-nitrogen codoping method. Phys Status Solidi B, 2013, 250:2102 https://www.researchgate.net/publication/264251371_Stable_p-type_ZnO_films_obtained_by_lithium-nitrogen_codoping_method
|
| [12] |
Park C H, Zhang S B, Wei S H. Origin of p-type doping difficulty in ZnO:the impurity perspective. Phys Rev B, 2002, 66:073202 doi: 10.1103/PhysRevB.66.073202
|
| [13] |
Yang X P, Lu J G, Zhang H H, et al. Preparation and XRD analyses of Na-doped ZnO nanorod arrays based on experiment and theory. Chem Phys Lett, 2012, 528(3):16 https://www.researchgate.net/publication/256683125_Preparation_and_XRD_Analyses_of_Na-Doped_ZnO_Nanorod_Arrays_Based_on_Experiment_and_Theory
|
| [14] |
Lin S S, Lu J G, Ye Z Z, et al. p-type behavior in Na-doped ZnO films and ZnO homojunction light-emitting diodes. Solid State Commun, 2008, 148:25 doi: 10.1016/j.ssc.2008.07.028
|
| [15] |
Pandey S K, Awasthi V, Sengar B S, et al. Band alignment and photon extraction studies of Na-doped MgZnO/Ga-doped ZnO heterojunction for light-emitter applications. J Appl Phys, 2015, 118:165301 doi: 10.1063/1.4934560
|
| [16] |
Persson C, Platzer-Björkman C, Malmström J, et al. Strong valence-band offset bowing of ZnO1-xSx enhances p-type nitrogen doping of ZnO-like alloys. Phys Rev Lett, 2006, 97:146403 doi: 10.1103/PhysRevLett.97.146403
|
| [17] |
Xu T N, Li X, Lu Z, et al. Realization of Ag-S codoped p-type ZnO thin films. Appl Sur Sci, 2014, 316:62 doi: 10.1016/j.apsusc.2014.07.156
|
| [18] |
Xu Y, Yang T, Yao B, et al. Influence of Ag-S codoping on silver chemical states and stable p-type conduction behavior of the ZnO films. Ceram Int, 2014, 40:2161 doi: 10.1016/j.ceramint.2013.07.133
|
| [19] |
Niu W Z, Xu H B, Guo Y M, et al. The effect of sulfur on the electrical properties of S and N co-doped ZnO thin films:experiment and first-principles calculations. Phys Chem Chem Phys, 2015, 17:16705 doi: 10.1039/C5CP02434J
|
| [20] |
Perdew J P, Burke S, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett, 1996, 77:3865 doi: 10.1103/PhysRevLett.77.3865
|
| [21] |
Van de Walle C G, Neugebauer J. First-principles calculations for defects and impurities:applications to Ⅲ-nitrides. J Appl Phys, 2004, 95:3851 doi: 10.1063/1.1682673
|
| [22] |
Oba F, Togo A, Tanaka I, et al. Defect energetics in ZnO:a hybrid Hartree-Fock density functional study. Phys Rev B, 2008, 77:245202 doi: 10.1103/PhysRevB.77.245202
|
| [23] |
Yan Y F, Zhang S B, Pantelides S T. Control of doping by impurity chemical potentials:predictions for p-type ZnO. Phys Rev Lett, 2001, 86:5723 doi: 10.1103/PhysRevLett.86.5723
|
| [24] |
Guo T T, Dong G B, Chena Q, et al. First-principles calculation on p-type conduction of (Sb, N) codoping in ZnO. J Phys Chem Solid, 2014, 75:42 doi: 10.1016/j.jpcs.2013.08.006
|
| [25] |
Zhang J K, Deng S H, Jin H, et al. First-principle study on the electronic structure and p-type conductivity of ZnO. Acta Phys Sin, 2007, 56:5371 https://www.researchgate.net/publication/291645152_First-principle_study_on_the_electronic_structure_and_p-type_conductivity_of_ZnO
|
| [26] |
Limpijumnong S, Zhang S B, Wei S H, et al. Doping by large-size-mismatched impurities:the microscopic origin of arsenic-or antimony-doped p-type zinc oxide. Phys Rev Lett, 2004, 92:155504 doi: 10.1103/PhysRevLett.92.155504
|









Article views: 2969 Times PDF downloads: 35 Times Cited by: 0 Times
Received: 31 October 2016 Revised: 12 February 2017 Online: Published: 01 August 2017
| Citation: |
Xingyi Tan, Qiang Li, Yongdan Zhu. First-principles study of p-type ZnO by S-Na co-doping[J]. Journal of Semiconductors, 2017, 38(8): 083001. doi: 10.1088/1674-4926/38/8/083001
****
X Y Tan, Q Li, Y D Zhu. First-principles study of p-type ZnO by S-Na co-doping[J]. J. Semicond., 2017, 38(8): 083001. doi: 10.1088/1674-4926/38/8/083001.

|
| [1] |
Özgür Ü, Alivov Y I, Liu C, et al. A comprehensive review of ZnO materials and devices. J Appl Phys, 2005, 98:0413011 https://www.researchgate.net/publication/276379347_A_Comprehensive_Review_of_ZnO_Materials_and_Devices
|
| [2] |
Sara K G, Ramin Y, Farid J S, et al. Optical and electrical properties of p-type Ag-doped ZnO nanostructures. Ceram Int, 2014, 40:7957 doi: 10.1016/j.ceramint.2013.12.145
|
| [3] |
Li W J, Kong C Y, Qin G P, et al. p-type conductivity and stability of Ag-N codoped ZnO thin films. J. Alloys Compd, 2014, 609:173 doi: 10.1016/j.jallcom.2014.04.051
|
| [4] |
Alivov Y I, Kalinina E V, Cherenkov A E, et al. Fabrication and characterization of n-ZnO/p-AlGaN heterojunction light-emitting diodes on 6H-SiC substrates. Appl Phys Lett, 2003, 83:4719 doi: 10.1063/1.1632537
|
| [5] |
Huang Y B, Zhou W, Wu P. Room-temperature ferromagnetism in epitaxial p-type K-doped ZnO films. Solid State Commun, 2014, 183:31 doi: 10.1016/j.ssc.2013.12.024
|
| [6] |
Makino T, Chia C H, Tuan N T, et al. Radiative and nonradiative recombination processes in lattice-matched (Cd, Zn)O/(Mg, Zn)O multiquantum wells. Appl Phys Lett, 2000, 77:1632 doi: 10.1063/1.1308540
|
| [7] |
Sáaedi A, Yousefi R, Jamali-Sheini F, et al. Optical and electrical properties of p-type Li-doped ZnO nanowires. Superlattices Microstruct, 2013, 61:91 doi: 10.1016/j.spmi.2013.06.014
|
| [8] |
Zhang S B, Wei S H, Zunger A. Intrinsic n-type versus p-type doping asymmetry and the defect physics of ZnO. Phys Rev B, 2001, 63:075205 doi: 10.1103/PhysRevB.63.075205
|
| [9] |
Dhara S, Giri P K. Stable p-type conductivity and enhanced photoconductivity from nitrogen-doped annealed ZnO thin film. Thin Solid Films, 2012, 520:5000 doi: 10.1016/j.tsf.2012.02.081
|
| [10] |
Liu J S, Shan C X, Shen H, et al. ZnO light-emitting devices with a lifetime of 6.8 hours. Appl Phys Lett, 2012, 101:011106 doi: 10.1063/1.4733298
|
| [11] |
Shen H, Shan C X, Liu J S, et al. Stable p-type ZnO films obtained by lithium-nitrogen codoping method. Phys Status Solidi B, 2013, 250:2102 https://www.researchgate.net/publication/264251371_Stable_p-type_ZnO_films_obtained_by_lithium-nitrogen_codoping_method
|
| [12] |
Park C H, Zhang S B, Wei S H. Origin of p-type doping difficulty in ZnO:the impurity perspective. Phys Rev B, 2002, 66:073202 doi: 10.1103/PhysRevB.66.073202
|
| [13] |
Yang X P, Lu J G, Zhang H H, et al. Preparation and XRD analyses of Na-doped ZnO nanorod arrays based on experiment and theory. Chem Phys Lett, 2012, 528(3):16 https://www.researchgate.net/publication/256683125_Preparation_and_XRD_Analyses_of_Na-Doped_ZnO_Nanorod_Arrays_Based_on_Experiment_and_Theory
|
| [14] |
Lin S S, Lu J G, Ye Z Z, et al. p-type behavior in Na-doped ZnO films and ZnO homojunction light-emitting diodes. Solid State Commun, 2008, 148:25 doi: 10.1016/j.ssc.2008.07.028
|
| [15] |
Pandey S K, Awasthi V, Sengar B S, et al. Band alignment and photon extraction studies of Na-doped MgZnO/Ga-doped ZnO heterojunction for light-emitter applications. J Appl Phys, 2015, 118:165301 doi: 10.1063/1.4934560
|
| [16] |
Persson C, Platzer-Björkman C, Malmström J, et al. Strong valence-band offset bowing of ZnO1-xSx enhances p-type nitrogen doping of ZnO-like alloys. Phys Rev Lett, 2006, 97:146403 doi: 10.1103/PhysRevLett.97.146403
|
| [17] |
Xu T N, Li X, Lu Z, et al. Realization of Ag-S codoped p-type ZnO thin films. Appl Sur Sci, 2014, 316:62 doi: 10.1016/j.apsusc.2014.07.156
|
| [18] |
Xu Y, Yang T, Yao B, et al. Influence of Ag-S codoping on silver chemical states and stable p-type conduction behavior of the ZnO films. Ceram Int, 2014, 40:2161 doi: 10.1016/j.ceramint.2013.07.133
|
| [19] |
Niu W Z, Xu H B, Guo Y M, et al. The effect of sulfur on the electrical properties of S and N co-doped ZnO thin films:experiment and first-principles calculations. Phys Chem Chem Phys, 2015, 17:16705 doi: 10.1039/C5CP02434J
|
| [20] |
Perdew J P, Burke S, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett, 1996, 77:3865 doi: 10.1103/PhysRevLett.77.3865
|
| [21] |
Van de Walle C G, Neugebauer J. First-principles calculations for defects and impurities:applications to Ⅲ-nitrides. J Appl Phys, 2004, 95:3851 doi: 10.1063/1.1682673
|
| [22] |
Oba F, Togo A, Tanaka I, et al. Defect energetics in ZnO:a hybrid Hartree-Fock density functional study. Phys Rev B, 2008, 77:245202 doi: 10.1103/PhysRevB.77.245202
|
| [23] |
Yan Y F, Zhang S B, Pantelides S T. Control of doping by impurity chemical potentials:predictions for p-type ZnO. Phys Rev Lett, 2001, 86:5723 doi: 10.1103/PhysRevLett.86.5723
|
| [24] |
Guo T T, Dong G B, Chena Q, et al. First-principles calculation on p-type conduction of (Sb, N) codoping in ZnO. J Phys Chem Solid, 2014, 75:42 doi: 10.1016/j.jpcs.2013.08.006
|
| [25] |
Zhang J K, Deng S H, Jin H, et al. First-principle study on the electronic structure and p-type conductivity of ZnO. Acta Phys Sin, 2007, 56:5371 https://www.researchgate.net/publication/291645152_First-principle_study_on_the_electronic_structure_and_p-type_conductivity_of_ZnO
|
| [26] |
Limpijumnong S, Zhang S B, Wei S H, et al. Doping by large-size-mismatched impurities:the microscopic origin of arsenic-or antimony-doped p-type zinc oxide. Phys Rev Lett, 2004, 92:155504 doi: 10.1103/PhysRevLett.92.155504
|

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2

 DownLoad:
DownLoad:

