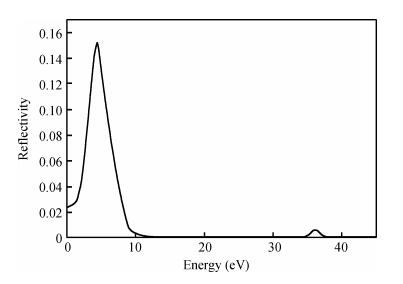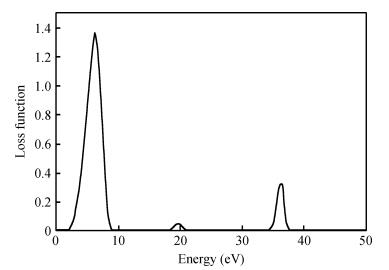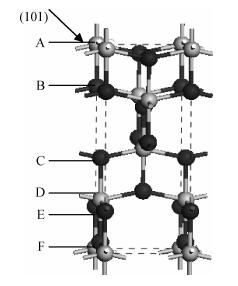
Fig. 1.
Direction of the different shear positions of the anatase TiO2 (101)
SEMICONDUCTOR MATERIALS
Ying Yang, Qing Feng, Weihua Wang and Yin Wang
Corresponding author: Feng Qing, Email:fengq_126@163.com
Abstract: The TiO2 (101) surface was studied using the plane-wave ultrasoft pseudopotential method based on the density functional theory, with emphasis on the structure, surface energy, band structure, density of states, and charge population. The anatase TiO2 (101) crystal surface structure, whose outermost and second layers were terminated by twofold coordinated oxygen atoms and fivefold coordinated titanium atoms, was found to be much more stable. The surface energy of the 18-layer atoms model was 0.580 J/m2. The surface electronic structure was similar to that of the bulk and no surface state. Compared with the bulk structure, the band gap increased 0.36 eV, the Ti5c-O2c bond lengths reduced 0.171 Å after relaxation, and the charges of the surface were transferred to the body. Analysis of the optical properties of the TiO2 (101) surface showed that it did not absorb in the low-energy region. An absorption edge in the ultraviolet region corresponding to the energy of 3.06 eV was found.
Key words: anatase, TiO2 (101) surface, first-principles, density functional theory, electronic structure
| [1] |
Tao Q, Wang J. Application of TiO2 photocatalytic oxidation technology to gas-phase contaminants degradation. Safety and Environmental Engineering, 2010, 17(5):26
|
| [2] |
Hebenstreit W, Ruzycki N, Herman G S, et al. Scanning tunneling microscopy investigation of the TiO2 anatase (101) surfaces. Phys Rev B, 2000, 62:16334 doi: 10.1103/PhysRevB.62.R16334
|
| [3] |
Hengerer R, Bolliger B, Erbudak M, et al. Structure and stability of the anatase TiO2 (101) and (001) surfaces. Surf Sci, 2000, 460:162 doi: 10.1016/S0039-6028(00)00527-6
|
| [4] |
Ma Xinguo, Tang Chaoqun, Huang Jinqiu, et al. First-principle calculations on the geometry and relaxation structure of anatase TiO2 (101) surface. Acta Physica Sinica, 2006, 55(8):4208
|
| [5] |
Ma Xinguo, Jiang Jianjun, Liang Pei. Theoretical study of native point defects on anatase TiO2 (101) surface. Acta Physica Sinica, 2008, 57:3120
|
| [6] |
Finazzi E, Valentin C D, Pacchioni A S G. First principles study of nitrogen doping at the anatase TiO2 (101) surface. Phys Chem C, 2007, 111(26):9275 doi: 10.1021/jp071186s
|
| [7] |
Chen Qili, Tang Chaoqun. First-principles calculations on electronic structures of N/F-doped and N-F-codoped TiO2 anatase (101) surfaces. Acta Physico-Chimica Sinica, 2009, 25(5):915
|
| [8] |
Chen Q, Tang C, Zheng G. First-principles study of TiO2 anatase (101) surfaces doped with N. Phys B, 2009, 404:1074 doi: 10.1016/j.physb.2008.11.032
|
| [9] |
Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B, 1999, 59:1758
|
| [10] |
Zhao Zongyan, Liu Qingju, Zhu Zhongqi, et al. First-principles calculation of electronic structure and optical properties of anatase TiO2. Chinese Journal of Semiconductors, 2007, 28(10):1555
|
| [11] |
Perdew J P, Chevary J A, Vosko S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation. Phys Rev B, 1992, 46:6671 doi: 10.1103/PhysRevB.46.6671
|
| [12] |
Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B, 1992, 45:13244 doi: 10.1103/PhysRevB.45.13244
|
| [13] |
Hammer B, Hansen L B, Norskov J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functional. Phys Rev B, 1999, 59:7413 doi: 10.1103/PhysRevB.59.7413
|
| [14] |
Wu Z, Cohen R E. A more accurate generalized gradient approximation for solids. Phys Rev B, 2006, 73:235116 doi: 10.1103/PhysRevB.73.235116
|
| [15] |
Perdew J P, Ruzsinszky A, Csonka G, et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett, 2008, 100:136406 doi: 10.1103/PhysRevLett.100.136406
|
| [16] |
Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method. Phys Rev Lett, 1980, 45:566 doi: 10.1103/PhysRevLett.45.566
|
| [17] |
Diebold U. The surface science of titanium dioxide. Surf Sci Rep, 2003, 48:53 doi: 10.1016/S0167-5729(02)00100-0
|
| [18] |
Segall M D, Lindan P L D, Probert M J, et al. First-principles simulation:ideas, illustrations and the CASTEP code. Journal of Physics:Condensed Matter, 2002, 14:2717 doi: 10.1088/0953-8984/14/11/301
|
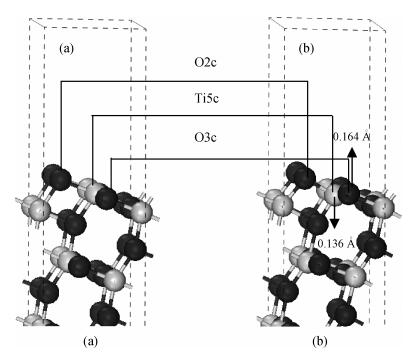
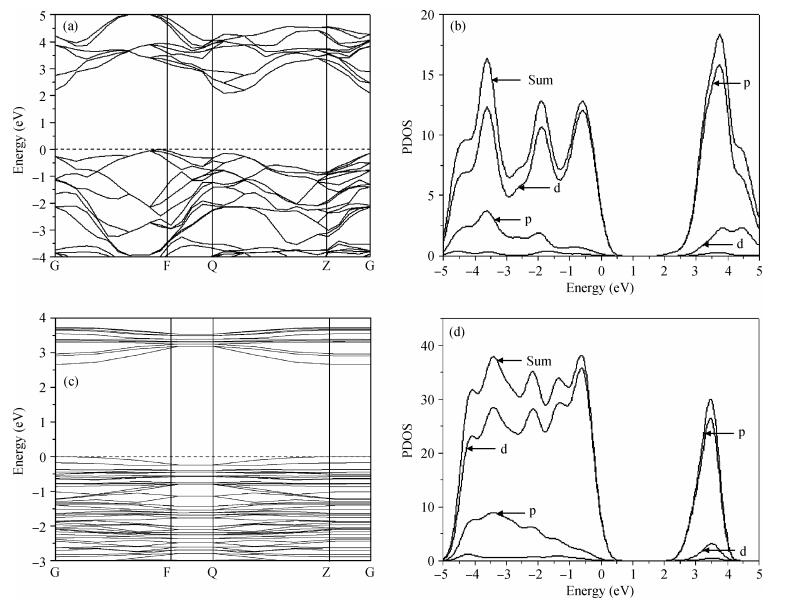
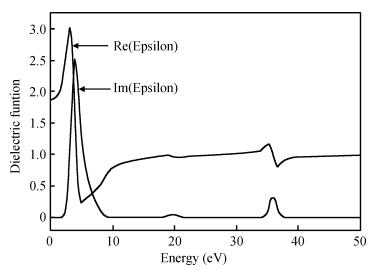
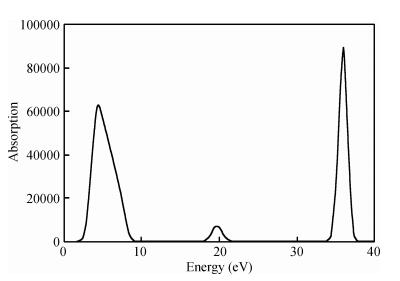
Table 1. Experimental and theoretical values of anatase TiO2 crystal parameters

|
Table 2. Total energy of different atomic termination structures with same atomic number

|
Table 3. Bond lengths and population of anatase TiO2 (101) surface before and after optimization

|
Table 4. Charge distribution of anatase TiO2 (101) surface

|
| [1] |
Tao Q, Wang J. Application of TiO2 photocatalytic oxidation technology to gas-phase contaminants degradation. Safety and Environmental Engineering, 2010, 17(5):26
|
| [2] |
Hebenstreit W, Ruzycki N, Herman G S, et al. Scanning tunneling microscopy investigation of the TiO2 anatase (101) surfaces. Phys Rev B, 2000, 62:16334 doi: 10.1103/PhysRevB.62.R16334
|
| [3] |
Hengerer R, Bolliger B, Erbudak M, et al. Structure and stability of the anatase TiO2 (101) and (001) surfaces. Surf Sci, 2000, 460:162 doi: 10.1016/S0039-6028(00)00527-6
|
| [4] |
Ma Xinguo, Tang Chaoqun, Huang Jinqiu, et al. First-principle calculations on the geometry and relaxation structure of anatase TiO2 (101) surface. Acta Physica Sinica, 2006, 55(8):4208
|
| [5] |
Ma Xinguo, Jiang Jianjun, Liang Pei. Theoretical study of native point defects on anatase TiO2 (101) surface. Acta Physica Sinica, 2008, 57:3120
|
| [6] |
Finazzi E, Valentin C D, Pacchioni A S G. First principles study of nitrogen doping at the anatase TiO2 (101) surface. Phys Chem C, 2007, 111(26):9275 doi: 10.1021/jp071186s
|
| [7] |
Chen Qili, Tang Chaoqun. First-principles calculations on electronic structures of N/F-doped and N-F-codoped TiO2 anatase (101) surfaces. Acta Physico-Chimica Sinica, 2009, 25(5):915
|
| [8] |
Chen Q, Tang C, Zheng G. First-principles study of TiO2 anatase (101) surfaces doped with N. Phys B, 2009, 404:1074 doi: 10.1016/j.physb.2008.11.032
|
| [9] |
Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B, 1999, 59:1758
|
| [10] |
Zhao Zongyan, Liu Qingju, Zhu Zhongqi, et al. First-principles calculation of electronic structure and optical properties of anatase TiO2. Chinese Journal of Semiconductors, 2007, 28(10):1555
|
| [11] |
Perdew J P, Chevary J A, Vosko S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation. Phys Rev B, 1992, 46:6671 doi: 10.1103/PhysRevB.46.6671
|
| [12] |
Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B, 1992, 45:13244 doi: 10.1103/PhysRevB.45.13244
|
| [13] |
Hammer B, Hansen L B, Norskov J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functional. Phys Rev B, 1999, 59:7413 doi: 10.1103/PhysRevB.59.7413
|
| [14] |
Wu Z, Cohen R E. A more accurate generalized gradient approximation for solids. Phys Rev B, 2006, 73:235116 doi: 10.1103/PhysRevB.73.235116
|
| [15] |
Perdew J P, Ruzsinszky A, Csonka G, et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett, 2008, 100:136406 doi: 10.1103/PhysRevLett.100.136406
|
| [16] |
Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method. Phys Rev Lett, 1980, 45:566 doi: 10.1103/PhysRevLett.45.566
|
| [17] |
Diebold U. The surface science of titanium dioxide. Surf Sci Rep, 2003, 48:53 doi: 10.1016/S0167-5729(02)00100-0
|
| [18] |
Segall M D, Lindan P L D, Probert M J, et al. First-principles simulation:ideas, illustrations and the CASTEP code. Journal of Physics:Condensed Matter, 2002, 14:2717 doi: 10.1088/0953-8984/14/11/301
|









Article views: 3401 Times PDF downloads: 59 Times Cited by: 0 Times
Received: 29 October 2012 Revised: 19 February 2013 Online: Published: 01 July 2013
| Citation: |
Ying Yang, Qing Feng, Weihua Wang, Yin Wang. First-principle study on the electronic and optical properties of the anatase TiO2 (101) surface[J]. Journal of Semiconductors, 2013, 34(7): 073004. doi: 10.1088/1674-4926/34/7/073004
****
Y Yang, Q Feng, W H Wang, Y Wang. First-principle study on the electronic and optical properties of the anatase TiO2 (101) surface[J]. J. Semicond., 2013, 34(7): 073004. doi: 10.1088/1674-4926/34/7/073004.

|
| [1] |
Tao Q, Wang J. Application of TiO2 photocatalytic oxidation technology to gas-phase contaminants degradation. Safety and Environmental Engineering, 2010, 17(5):26
|
| [2] |
Hebenstreit W, Ruzycki N, Herman G S, et al. Scanning tunneling microscopy investigation of the TiO2 anatase (101) surfaces. Phys Rev B, 2000, 62:16334 doi: 10.1103/PhysRevB.62.R16334
|
| [3] |
Hengerer R, Bolliger B, Erbudak M, et al. Structure and stability of the anatase TiO2 (101) and (001) surfaces. Surf Sci, 2000, 460:162 doi: 10.1016/S0039-6028(00)00527-6
|
| [4] |
Ma Xinguo, Tang Chaoqun, Huang Jinqiu, et al. First-principle calculations on the geometry and relaxation structure of anatase TiO2 (101) surface. Acta Physica Sinica, 2006, 55(8):4208
|
| [5] |
Ma Xinguo, Jiang Jianjun, Liang Pei. Theoretical study of native point defects on anatase TiO2 (101) surface. Acta Physica Sinica, 2008, 57:3120
|
| [6] |
Finazzi E, Valentin C D, Pacchioni A S G. First principles study of nitrogen doping at the anatase TiO2 (101) surface. Phys Chem C, 2007, 111(26):9275 doi: 10.1021/jp071186s
|
| [7] |
Chen Qili, Tang Chaoqun. First-principles calculations on electronic structures of N/F-doped and N-F-codoped TiO2 anatase (101) surfaces. Acta Physico-Chimica Sinica, 2009, 25(5):915
|
| [8] |
Chen Q, Tang C, Zheng G. First-principles study of TiO2 anatase (101) surfaces doped with N. Phys B, 2009, 404:1074 doi: 10.1016/j.physb.2008.11.032
|
| [9] |
Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B, 1999, 59:1758
|
| [10] |
Zhao Zongyan, Liu Qingju, Zhu Zhongqi, et al. First-principles calculation of electronic structure and optical properties of anatase TiO2. Chinese Journal of Semiconductors, 2007, 28(10):1555
|
| [11] |
Perdew J P, Chevary J A, Vosko S H, et al. Atoms, molecules, solids, and surfaces:applications of the generalized gradient approximation for exchange and correlation. Phys Rev B, 1992, 46:6671 doi: 10.1103/PhysRevB.46.6671
|
| [12] |
Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B, 1992, 45:13244 doi: 10.1103/PhysRevB.45.13244
|
| [13] |
Hammer B, Hansen L B, Norskov J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functional. Phys Rev B, 1999, 59:7413 doi: 10.1103/PhysRevB.59.7413
|
| [14] |
Wu Z, Cohen R E. A more accurate generalized gradient approximation for solids. Phys Rev B, 2006, 73:235116 doi: 10.1103/PhysRevB.73.235116
|
| [15] |
Perdew J P, Ruzsinszky A, Csonka G, et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys Rev Lett, 2008, 100:136406 doi: 10.1103/PhysRevLett.100.136406
|
| [16] |
Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method. Phys Rev Lett, 1980, 45:566 doi: 10.1103/PhysRevLett.45.566
|
| [17] |
Diebold U. The surface science of titanium dioxide. Surf Sci Rep, 2003, 48:53 doi: 10.1016/S0167-5729(02)00100-0
|
| [18] |
Segall M D, Lindan P L D, Probert M J, et al. First-principles simulation:ideas, illustrations and the CASTEP code. Journal of Physics:Condensed Matter, 2002, 14:2717 doi: 10.1088/0953-8984/14/11/301
|

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2

 DownLoad:
DownLoad: