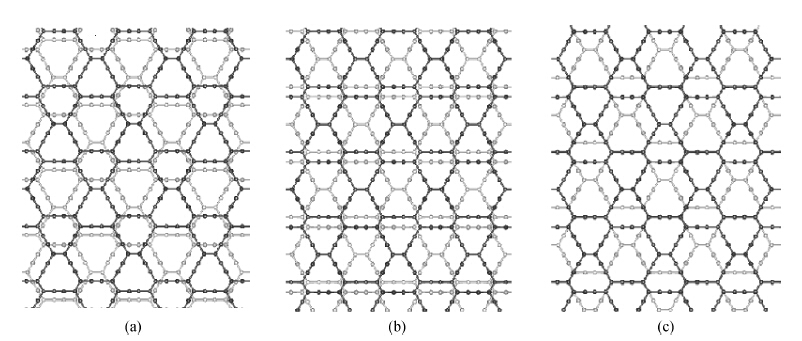
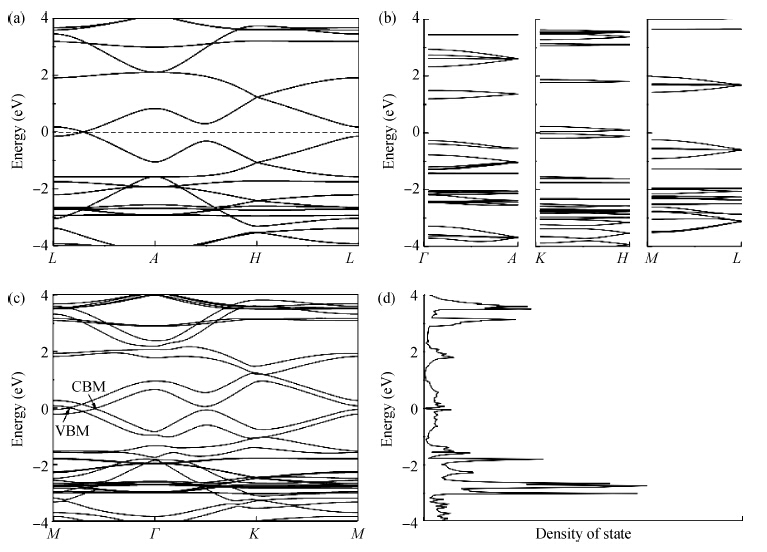
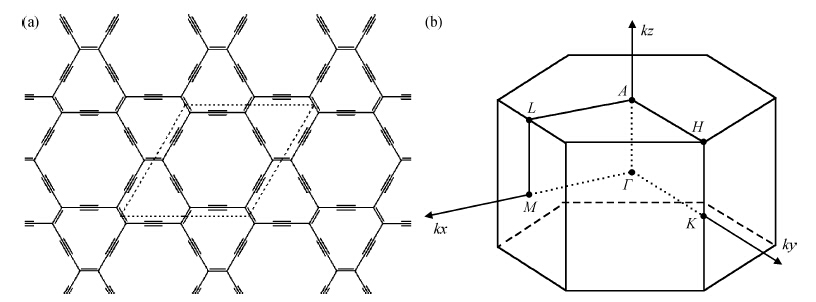
β-graphyne, a carbon allotrope, is a gapless semiconductor with hexagonal lattice symmetry, just like graphene. We calculated the optimized structure and electronic structures of some possible three-dimensional
β-graphyne stacking arrangements by means of the first-principles frozen-core projector augmented-wave method implemented in the Vienna
ab initio simulation package. The optimized lattice constant
a of the three-dimensional
β-graphyne turns out to be 9.46 Å, which is slightly smaller than its two-dimensional counterpart. The binding energy is about 90% of that of graphite, which suggests that three-dimensional
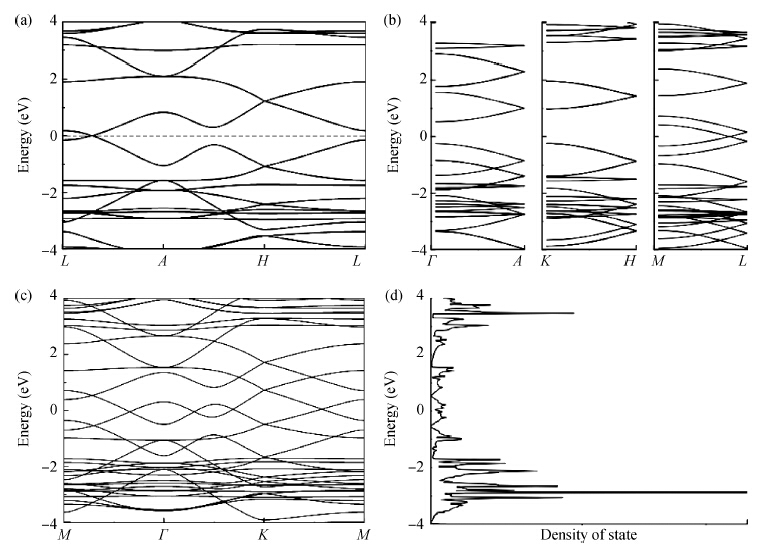
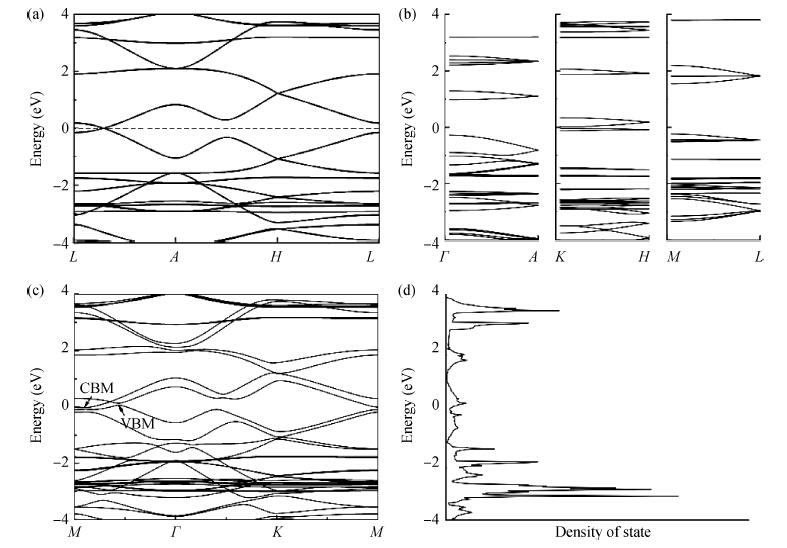
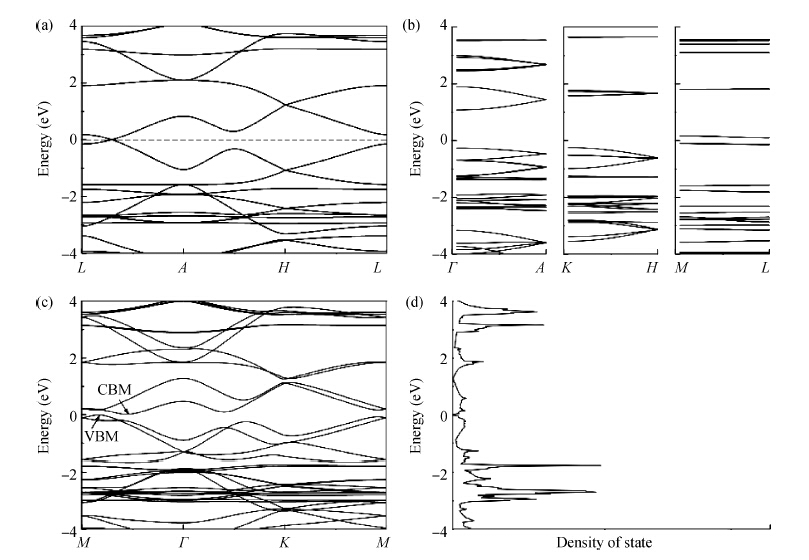
β-graphyne will be stable when it is synthesized. The band structure is calculated via the hybrid functional. We found that the most stable three-dimensional stacking arrangement is an indirect band gap semiconductor with an energy gap of 0.1 eV.



 DownLoad:
DownLoad: