


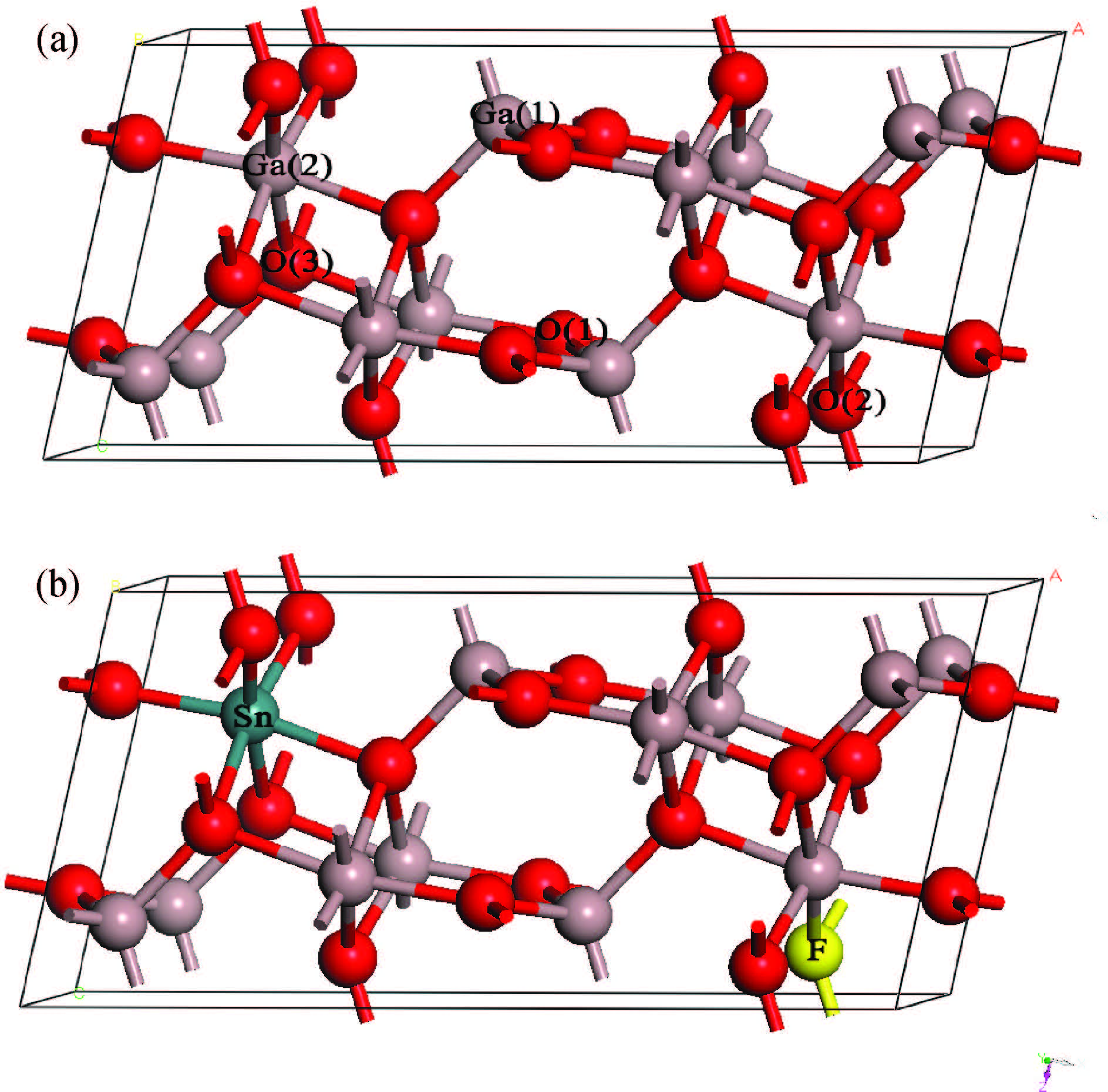
Fig1.
{(Color online) Crystal structures of (a) β -Ga2O3 and (b) Sn/F co-doped β -Ga2O3.
SEMICONDUCTOR PHYSICS
Yinnü Zhao1, and Jinliang Yan2
Corresponding author: Zhao Yinnü, zhaoyinnv@sina.com
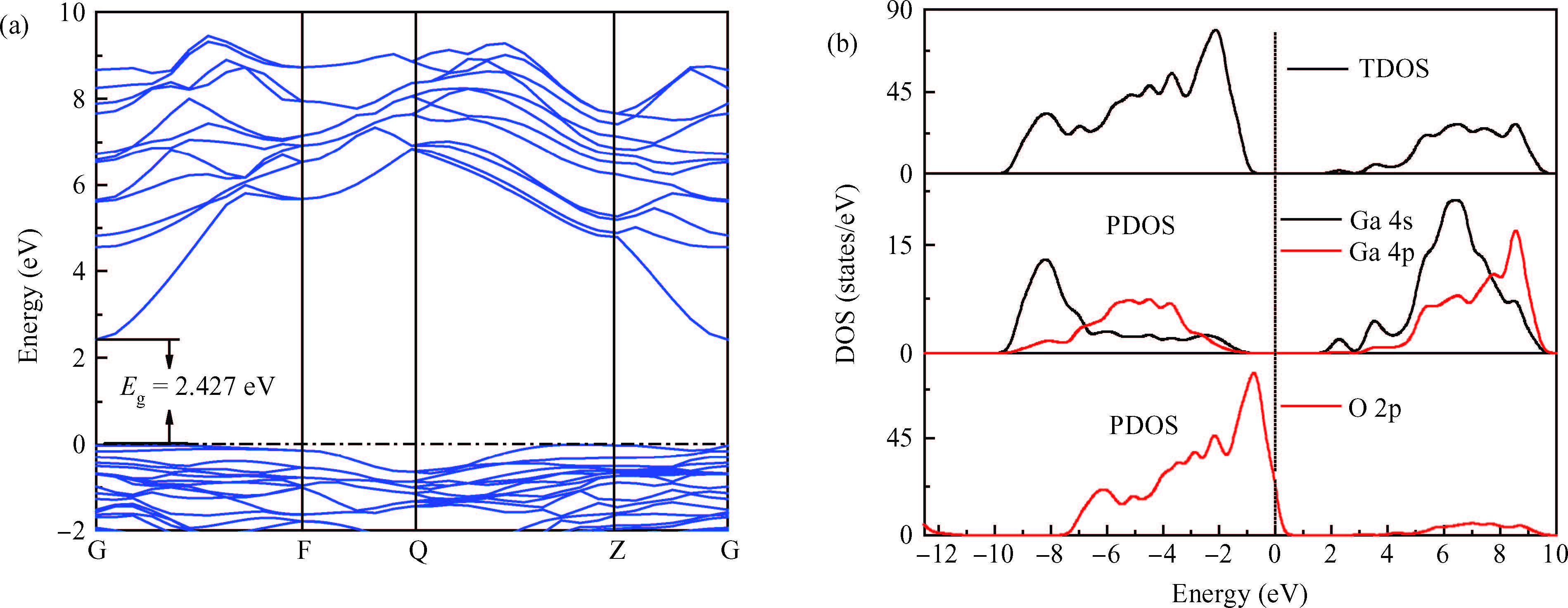
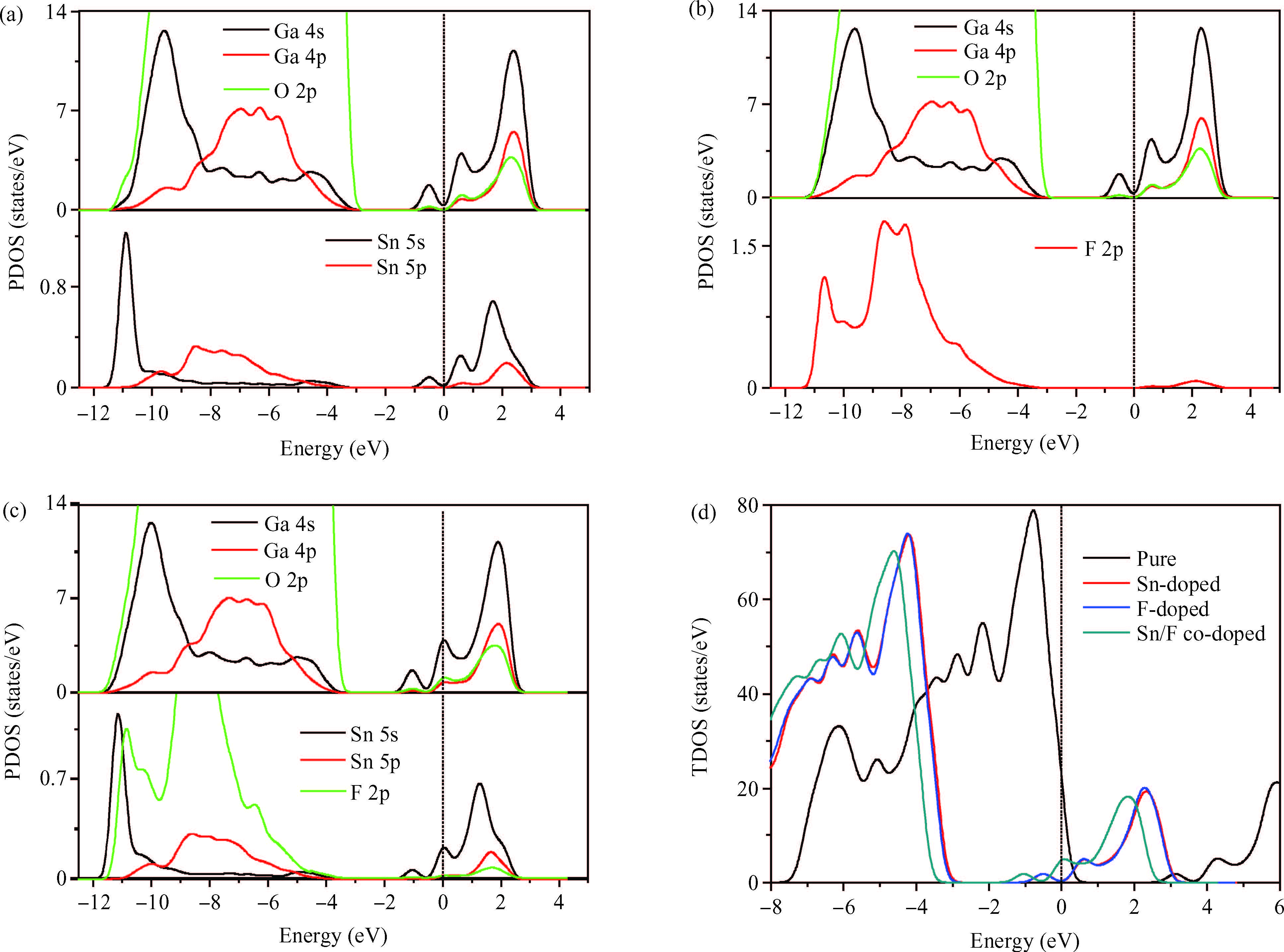
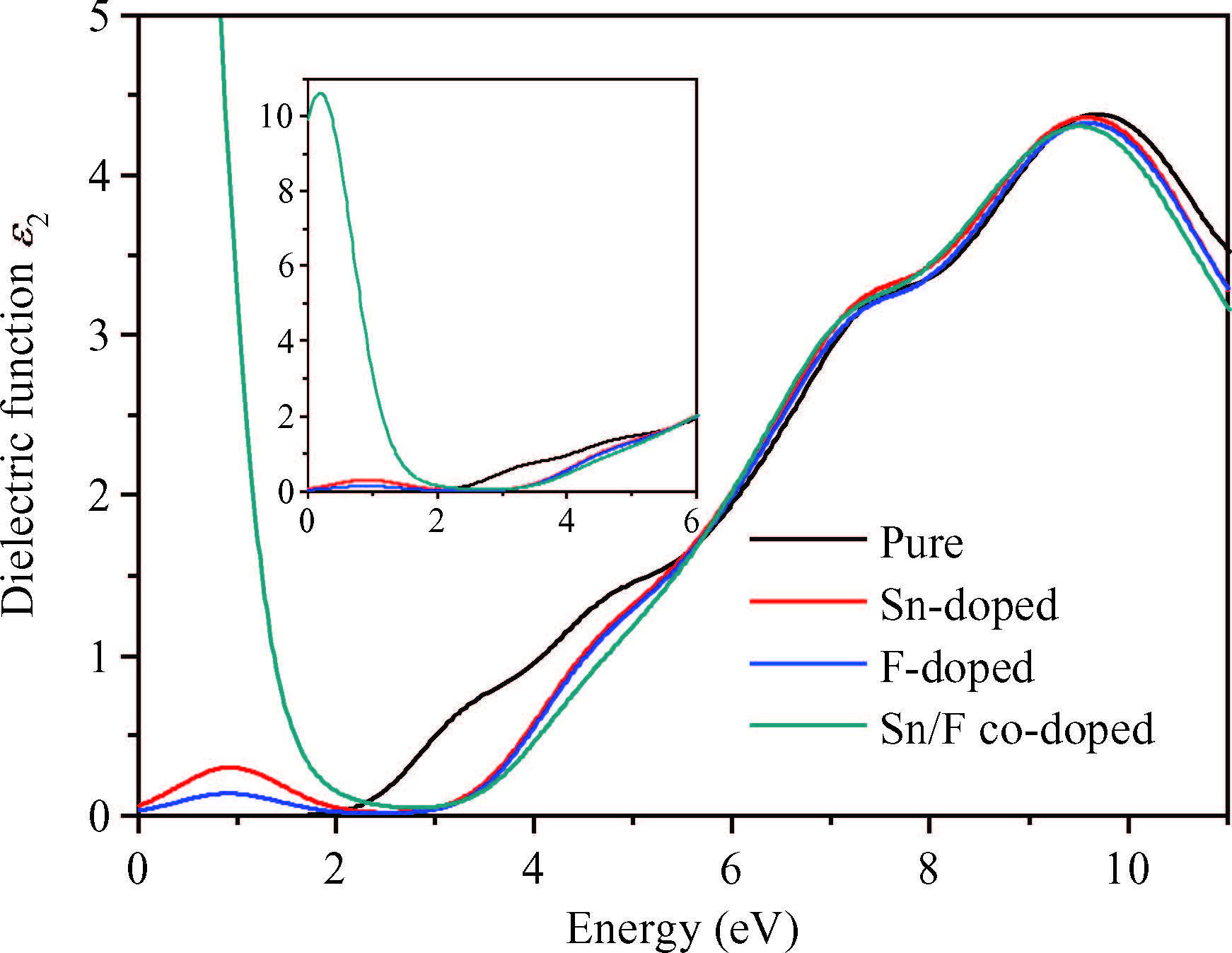
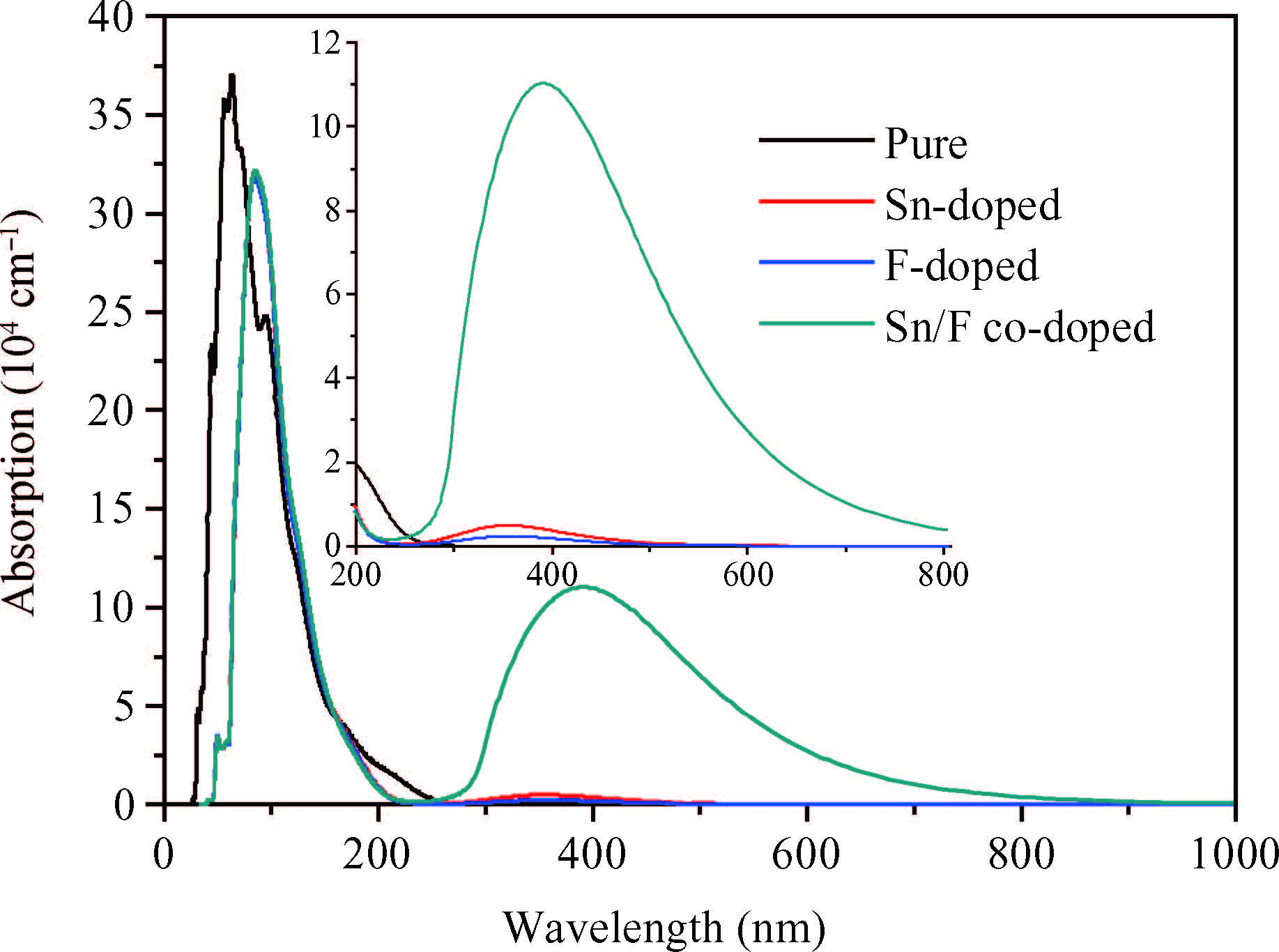
Abstract: Defect formation energies, electronic structures and optical properties of Sn-doped β-Ga2O3, F-doped β-Ga2O3, and Sn/F co-doped β-Ga2O3 were calculated using the first-principles. The calculated results of the pure and Sn-doped β-Ga2O3 using the local-density approximation (LDA) method show that the lattice parameters and electronic structures are in agreement with previous data. The defect formation energies demonstrate that the doped systems are relatively easy to form under O-rich conditions. Sn-doping, F-doping and Sn/F co-doping make β-Ga2O3 become an n-type semiconductor. Sn/F co-doping β-Ga2O3 has the smallest effective electron mass and the biggest relative electron number, which is expected to possess good conductivity. Sn/F co-doping β-Ga2O3 displays an intense absorption in visible light.
Keywords: semiconductor doping, electric properties, optical properties, electronic structure
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] |
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] |









Article views: 4383 Times PDF downloads: 77 Times Cited by: 0 Times
Received: 01 January 2015 Revised: Online: Published: 01 August 2015
| Citation: |
Yinnü Zhao, Jinliang Yan. First-principles study of n-type tin/fluorine co-doped beta-gallium oxides[J]. Journal of Semiconductors, 2015, 36(8): 082004. doi: 10.1088/1674-4926/36/8/082004
****
Y Zhao, J L Yan. First-principles study of n-type tin/fluorine co-doped beta-gallium oxides[J]. J. Semicond., 2015, 36(8): 082004. doi: 10.1088/1674-4926/36/8/082004.

|
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] |

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2

 DownLoad:
DownLoad:

