


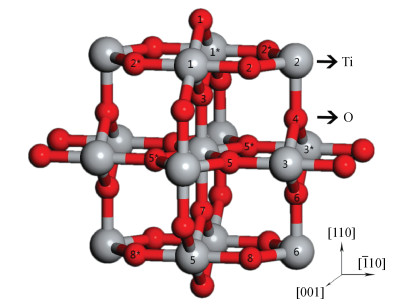
Fig. 1.
(Color online) Atomic label of the 3-trilayer slab. The larger and smaller balls are titanium and oxygen atoms, respectively. Symmetric atoms are marked with an asterisk (*).
SEMICONDUCTOR PHYSICS
Xuechao Li, Jianhao Shi and Rundong Wan
Corresponding author: Wan Rundong, Email:rdwan@kmust.edu.cn
Abstract: Traditionally, we use a slab to mimic a surface and we constrain the slab to have the bulk-terminated 2D lattice constants. Here we propose a different model in which we impose no constraints, allowing all coordinates including the 2D slab itself to relax. We perform DFT calculations on both models. We find that the unconstrained slabs yield better agreement with experimental results and they have lower total energies. The optimized 2D lattice constants of the unconstrained slabs eventually converge to the attached bulk value. The total energy difference reveals that, with odd number trilayers, the unconstrained slab is much closer to the corresponding constrained slab. The surface energies both converge to the individual values with the number of atomic layers.
Keywords: rutile TiO2, slab, DFT, LDA/6-31G
| [1] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [2] |
Pang C L, Lindsay R, Thornton G. Chemical reactions on rutile TiO2 (110). Chem Soc Rev, 2008, 37(10): 2328 doi: 10.1039/b719085a
|
| [3] |
Harrison N M, Wang X G, Muscat J, et al. The influence of soft vibrational modes on our understanding of oxide surface structure. Faraday Discussions, 1999, 114: 305 doi: 10.1039/a906386b
|
| [4] |
Zhang Y, Lin W, Li Y, et al. A theoretical study on the electronic structures of TiO2: effect of Hartree-Fock exchange. J Phys Chem B, 2005, 109(41): 19270 doi: 10.1021/jp0523625
|
| [5] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theory Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [6] |
Charlton G, Howes P B, Nicklin C L, et al. Relaxation of TiO2 (110)-(1×1) using surface X-ray diffraction. Physl Rev Lett, 1997, 78(3): 495 doi: 10.1103/PhysRevLett.78.495
|
| [7] |
Hebenstreit E L D, Hebenstreit W, Diebold U. Structures of sulfur on TiO2 (110) determined by scanning tunneling microscopy, X-ray photoelectron spectroscopy and low-energy electron diffraction. Surf Sci, 2001, 470(3): 347 doi: 10.1016/S0039-6028(00)00849-9
|
| [8] |
Bennett R A, Pang C L, Perkins N, et al. Surface structures in the SMSI state; Pd on (1×2) reconstructed TiO2 (110). J Phys Chem B, 2002, 106(18): 4688 doi: 10.1021/jp0138328
|
| [9] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [10] |
Pillay D, Hwang G S. Structure of small Aun, Agn, and Cun clusters (n=2-4) on rutile TiO2 (110): a density functional theory study. Journal of Molecular Structure: THEOCHEM, 2006, 771(1): 129
|
| [11] |
Perron H, Domain C, Roques J, et al. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theoretical Chemistry Accounts, 2007, 117(4): 565 doi: 10.1007/s00214-006-0189-y
|
| [12] |
Busayaporn W. TiO2 (110) surface structure. University of Manchester, 2010
|
| [13] |
Lindsay R, Wander A, Ernst A, et al. Revisiting the surface structure of TiO2 (110): a quantitative low-energy electron diffraction study. Phys Rev Lett, 2005, 94(24): 246102 doi: 10.1103/PhysRevLett.94.246102
|
| [14] |
Cabailh G, Torrelles X, Lindsay R, et al. Geometric structure of TiO2 (110) (1×1): achieving experimental consensus. Phys Rev B, 2007, 75(24): 241403 doi: 10.1103/PhysRevB.75.241403
|
| [15] |
Busayaporn W, Torrelles X, Wander A, et al. Geometric structure of TiO2 (110) (1×1): confirming experimental conclusions. Phys Rev B, 2010, 81(15): 153404 doi: 10.1103/PhysRevB.81.153404
|
| [16] |
Ramamoorthy M, Vanderbilt D, King-Smith R D. First-principles calculations of the energetics of stoichiometric TiO2 surfaces. Phys Rev B, 1994, 49(23): 16721 doi: 10.1103/PhysRevB.49.16721
|
| [17] |
Bates S P, Kresse G, Gillan M J. A systematic study of the surface energetics and structure of TiO2 (110) by first-principles calculations. Surf Sci, 1997, 385(2): 386
|
| [18] |
Elliott S D, Bates S P. A first principles survey of stoichiometric (1×2) reconstructions on the rutile surface. Surf Sci, 2001, 495(3): 211 doi: 10.1016/S0039-6028(01)01328-0
|
| [19] |
Swamy V, Muscat J, Gale J D, et al. Simulation of low index rutile surfaces with a transferable variable-charge Ti-O interatomic potential and comparison with ab initio results. Surf Sci, 2002, 504: 115 doi: 10.1016/S0039-6028(01)01925-2
|
| [20] |
Vijay A, Mills G, Metiu H. Adsorption of gold on stoichiometric and reduced rutile TiO2 (110) surfaces. J Chem Phys, 2003, 118(14): 6536 doi: 10.1063/1.1557919
|
| [21] |
Wang Y, Hwang G S. Adsorption of Au atoms on stoichiometric and reduced TiO2 (110) rutile surfaces: a first principles study. Surf Sci, 2003, 542(1): 72
|
| [22] |
Sano H, Mizutani G, Wolf W, et al. Ab initio calculation of optical second harmonic generation at the rutile TiO2 (110) surface. Phys Rev B, 2004, 70(12): 125411 doi: 10.1103/PhysRevB.70.125411
|
| [23] |
Thompson S J, Lewis S P. Revisiting the (110) surface structure of TiO2: a theoretical analysis. Phys Rev B, 2006, 73(7): 073403 doi: 10.1103/PhysRevB.73.073403
|
| [24] |
Hameeuw K J, Cantele G, Ninno D, et al. The rutile TiO2 (110) surface: Obtaining converged structural properties from first-principles calculations. J Chem Phys, 2006, 124(2): 024708 doi: 10.1063/1.2136158
|
| [25] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theor Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [26] |
Thompson S J, Lewis S P. Deconstructing the structural convergence of the (110) surface of rutile TiO2. In: Computer simulation studies in condensed-matter physics XIX. Springer Berlin Heidelberg, 2009, 123: 26
|
| [27] |
Slater J C. The self-consistent field for molecules and solids. New York: McGraw-Hill, 1974
|
| [28] |
Vosko S H, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Canadian J Phys, 1980, 58(8): 1200 doi: 10.1139/p80-159
|
| [29] |
Binkley J S, Pople J A, Hehre W J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J Amer Chem Soc, 1980, 102(3): 939 doi: 10.1021/ja00523a008
|
| [30] |
Dobbs K D, Hehre W J. Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals. J Comput Chem, 1987, 8(6): 861 doi: 10.1002/(ISSN)1096-987X
|
| [31] |
Mattsson A, Hu S, Hermansson K, et al. Adsorption of formic acid on rutile TiO2 (110) revisited: an infrared reflection-absorption spectroscopy and density functional theory study. J Chem Phys, 2014, 140(3): 034705 doi: 10.1063/1.4855176
|
| [32] |
Chang T Y, Tanaka Y, Ishikawa R, et al. Direct imaging of Pt single atoms adsorbed on TiO2 (110) surfaces. Nano Lett, 2013, 14(1): 134
|
| [33] |
Rangan S, Ruggieri C, Bartynski R, et al. Densely packed ZnTPPs monolayer on the rutile TiO2 (110)-(1×1) surface: adsorption behavior and energy level alignment. J Phys Chem C, 2016, 120(8): 4430 doi: 10.1021/acs.jpcc.5b12736
|
| [34] |
Ong S V, Khanna S N. Theoretical studies of the stability and oxidation of Pdn (n=1-7) clusters on rutile TiO2 (110): adsorption on the stoichiometric surface. J Phys Chem C, 2012, 116(4): 3105 doi: 10.1021/jp212504x
|
| [35] |
Zhang Z, Tang M, Wang Z T, et al. Imaging of formaldehyde adsorption and diffusion on TiO2 (110). Topics in Catalysis, 2015, 58(2/3): 103
|
| [36] |
García-Mota M, Vojvodic A, Abild-Pedersen F, et al. Electronic origin of the surface reactivity of transition-metal-doped TiO2 (110). J Phys Chem C, 2012, 117(1): 460
|
| [37] |
Sasahara A, Tomitori M. XPS and STM study of Nb-doped TiO2 (110)-(1×1) surfaces. J Phys Chem C, 2013, 117(34): 17680 doi: 10.1021/jp4057576
|
| [38] |
Frisch M, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision C. 01, Gaussian. Inc., Wallingford, CT, 2009
|
| [39] |
Materials Studio version 4.4 from Accelrys http://accelrys.com/events/webinars/materials-studio-44/
|
| [40] |
Heyd J, Scuseria G E. Efficient hybrid density functional calculations in solids: assessment of the Heyd-Scuseria-Ernzerh of screened Coulomb hybrid functional. J Chem Phys, 2004, 121(3): 1187 doi: 10.1063/1.1760074
|
| [41] |
Bredow T, Giordano L, Cinquini F, et al. Electronic properties of rutile TiO2 ultrathin films: odd-even oscillations with the number of layers. Phys Rev B, 2004, 70(3): 035419 doi: 10.1103/PhysRevB.70.035419
|
| [42] |
Evarestov R A, Bandura A V. Hartree-Fock calculations of electronic structure of (110)-surface of rutile TiO2: comparison of single (2D) and periodic (3D) slab models. International Journal of Quantum Chemistry, 2004, 96(3): 282 doi: 10.1002/(ISSN)1097-461X
|
| [43] |
Scaranto J, Mallia G, Giorgianni S, et al. A quantum-mechanical study of the vinyl fluoride adsorbed on the rutile TiO2 (110) surface. Surf Sci, 2006, 600(2): 305 doi: 10.1016/j.susc.2005.10.032
|
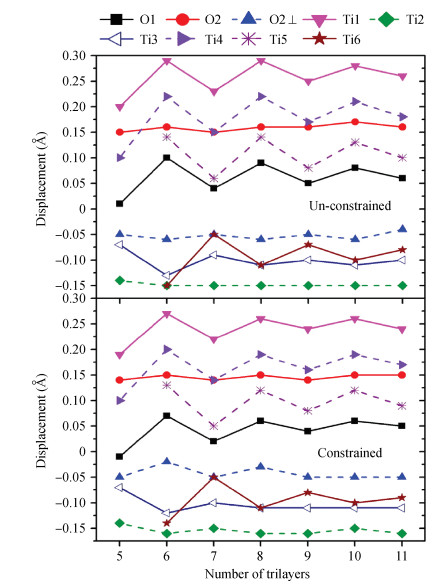
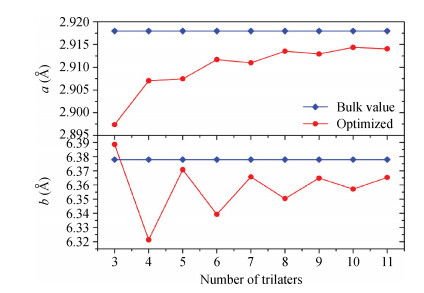
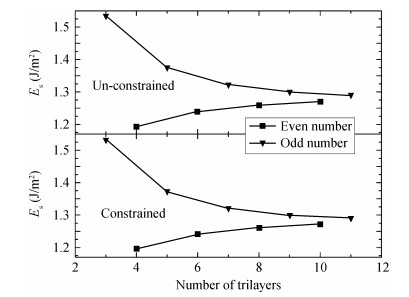
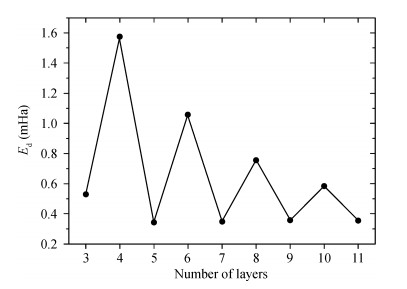
Table 1. Displacement comparisons between the different methods in Å, where UC denotes the unconstrained slabs and C denotes the constrained slabs, respectively. The displacements are along the [110] direction, except the lateral ones with the perpendicular signs in the [110] direction. LEED-IV and SXRD mean the atomic displacements obtained from the analysis of low-energy electron diffraction data and surface X-ray diffraction data in Reference [15].
 |
| [1] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [2] |
Pang C L, Lindsay R, Thornton G. Chemical reactions on rutile TiO2 (110). Chem Soc Rev, 2008, 37(10): 2328 doi: 10.1039/b719085a
|
| [3] |
Harrison N M, Wang X G, Muscat J, et al. The influence of soft vibrational modes on our understanding of oxide surface structure. Faraday Discussions, 1999, 114: 305 doi: 10.1039/a906386b
|
| [4] |
Zhang Y, Lin W, Li Y, et al. A theoretical study on the electronic structures of TiO2: effect of Hartree-Fock exchange. J Phys Chem B, 2005, 109(41): 19270 doi: 10.1021/jp0523625
|
| [5] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theory Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [6] |
Charlton G, Howes P B, Nicklin C L, et al. Relaxation of TiO2 (110)-(1×1) using surface X-ray diffraction. Physl Rev Lett, 1997, 78(3): 495 doi: 10.1103/PhysRevLett.78.495
|
| [7] |
Hebenstreit E L D, Hebenstreit W, Diebold U. Structures of sulfur on TiO2 (110) determined by scanning tunneling microscopy, X-ray photoelectron spectroscopy and low-energy electron diffraction. Surf Sci, 2001, 470(3): 347 doi: 10.1016/S0039-6028(00)00849-9
|
| [8] |
Bennett R A, Pang C L, Perkins N, et al. Surface structures in the SMSI state; Pd on (1×2) reconstructed TiO2 (110). J Phys Chem B, 2002, 106(18): 4688 doi: 10.1021/jp0138328
|
| [9] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [10] |
Pillay D, Hwang G S. Structure of small Aun, Agn, and Cun clusters (n=2-4) on rutile TiO2 (110): a density functional theory study. Journal of Molecular Structure: THEOCHEM, 2006, 771(1): 129
|
| [11] |
Perron H, Domain C, Roques J, et al. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theoretical Chemistry Accounts, 2007, 117(4): 565 doi: 10.1007/s00214-006-0189-y
|
| [12] |
Busayaporn W. TiO2 (110) surface structure. University of Manchester, 2010
|
| [13] |
Lindsay R, Wander A, Ernst A, et al. Revisiting the surface structure of TiO2 (110): a quantitative low-energy electron diffraction study. Phys Rev Lett, 2005, 94(24): 246102 doi: 10.1103/PhysRevLett.94.246102
|
| [14] |
Cabailh G, Torrelles X, Lindsay R, et al. Geometric structure of TiO2 (110) (1×1): achieving experimental consensus. Phys Rev B, 2007, 75(24): 241403 doi: 10.1103/PhysRevB.75.241403
|
| [15] |
Busayaporn W, Torrelles X, Wander A, et al. Geometric structure of TiO2 (110) (1×1): confirming experimental conclusions. Phys Rev B, 2010, 81(15): 153404 doi: 10.1103/PhysRevB.81.153404
|
| [16] |
Ramamoorthy M, Vanderbilt D, King-Smith R D. First-principles calculations of the energetics of stoichiometric TiO2 surfaces. Phys Rev B, 1994, 49(23): 16721 doi: 10.1103/PhysRevB.49.16721
|
| [17] |
Bates S P, Kresse G, Gillan M J. A systematic study of the surface energetics and structure of TiO2 (110) by first-principles calculations. Surf Sci, 1997, 385(2): 386
|
| [18] |
Elliott S D, Bates S P. A first principles survey of stoichiometric (1×2) reconstructions on the rutile surface. Surf Sci, 2001, 495(3): 211 doi: 10.1016/S0039-6028(01)01328-0
|
| [19] |
Swamy V, Muscat J, Gale J D, et al. Simulation of low index rutile surfaces with a transferable variable-charge Ti-O interatomic potential and comparison with ab initio results. Surf Sci, 2002, 504: 115 doi: 10.1016/S0039-6028(01)01925-2
|
| [20] |
Vijay A, Mills G, Metiu H. Adsorption of gold on stoichiometric and reduced rutile TiO2 (110) surfaces. J Chem Phys, 2003, 118(14): 6536 doi: 10.1063/1.1557919
|
| [21] |
Wang Y, Hwang G S. Adsorption of Au atoms on stoichiometric and reduced TiO2 (110) rutile surfaces: a first principles study. Surf Sci, 2003, 542(1): 72
|
| [22] |
Sano H, Mizutani G, Wolf W, et al. Ab initio calculation of optical second harmonic generation at the rutile TiO2 (110) surface. Phys Rev B, 2004, 70(12): 125411 doi: 10.1103/PhysRevB.70.125411
|
| [23] |
Thompson S J, Lewis S P. Revisiting the (110) surface structure of TiO2: a theoretical analysis. Phys Rev B, 2006, 73(7): 073403 doi: 10.1103/PhysRevB.73.073403
|
| [24] |
Hameeuw K J, Cantele G, Ninno D, et al. The rutile TiO2 (110) surface: Obtaining converged structural properties from first-principles calculations. J Chem Phys, 2006, 124(2): 024708 doi: 10.1063/1.2136158
|
| [25] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theor Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [26] |
Thompson S J, Lewis S P. Deconstructing the structural convergence of the (110) surface of rutile TiO2. In: Computer simulation studies in condensed-matter physics XIX. Springer Berlin Heidelberg, 2009, 123: 26
|
| [27] |
Slater J C. The self-consistent field for molecules and solids. New York: McGraw-Hill, 1974
|
| [28] |
Vosko S H, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Canadian J Phys, 1980, 58(8): 1200 doi: 10.1139/p80-159
|
| [29] |
Binkley J S, Pople J A, Hehre W J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J Amer Chem Soc, 1980, 102(3): 939 doi: 10.1021/ja00523a008
|
| [30] |
Dobbs K D, Hehre W J. Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals. J Comput Chem, 1987, 8(6): 861 doi: 10.1002/(ISSN)1096-987X
|
| [31] |
Mattsson A, Hu S, Hermansson K, et al. Adsorption of formic acid on rutile TiO2 (110) revisited: an infrared reflection-absorption spectroscopy and density functional theory study. J Chem Phys, 2014, 140(3): 034705 doi: 10.1063/1.4855176
|
| [32] |
Chang T Y, Tanaka Y, Ishikawa R, et al. Direct imaging of Pt single atoms adsorbed on TiO2 (110) surfaces. Nano Lett, 2013, 14(1): 134
|
| [33] |
Rangan S, Ruggieri C, Bartynski R, et al. Densely packed ZnTPPs monolayer on the rutile TiO2 (110)-(1×1) surface: adsorption behavior and energy level alignment. J Phys Chem C, 2016, 120(8): 4430 doi: 10.1021/acs.jpcc.5b12736
|
| [34] |
Ong S V, Khanna S N. Theoretical studies of the stability and oxidation of Pdn (n=1-7) clusters on rutile TiO2 (110): adsorption on the stoichiometric surface. J Phys Chem C, 2012, 116(4): 3105 doi: 10.1021/jp212504x
|
| [35] |
Zhang Z, Tang M, Wang Z T, et al. Imaging of formaldehyde adsorption and diffusion on TiO2 (110). Topics in Catalysis, 2015, 58(2/3): 103
|
| [36] |
García-Mota M, Vojvodic A, Abild-Pedersen F, et al. Electronic origin of the surface reactivity of transition-metal-doped TiO2 (110). J Phys Chem C, 2012, 117(1): 460
|
| [37] |
Sasahara A, Tomitori M. XPS and STM study of Nb-doped TiO2 (110)-(1×1) surfaces. J Phys Chem C, 2013, 117(34): 17680 doi: 10.1021/jp4057576
|
| [38] |
Frisch M, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision C. 01, Gaussian. Inc., Wallingford, CT, 2009
|
| [39] |
Materials Studio version 4.4 from Accelrys http://accelrys.com/events/webinars/materials-studio-44/
|
| [40] |
Heyd J, Scuseria G E. Efficient hybrid density functional calculations in solids: assessment of the Heyd-Scuseria-Ernzerh of screened Coulomb hybrid functional. J Chem Phys, 2004, 121(3): 1187 doi: 10.1063/1.1760074
|
| [41] |
Bredow T, Giordano L, Cinquini F, et al. Electronic properties of rutile TiO2 ultrathin films: odd-even oscillations with the number of layers. Phys Rev B, 2004, 70(3): 035419 doi: 10.1103/PhysRevB.70.035419
|
| [42] |
Evarestov R A, Bandura A V. Hartree-Fock calculations of electronic structure of (110)-surface of rutile TiO2: comparison of single (2D) and periodic (3D) slab models. International Journal of Quantum Chemistry, 2004, 96(3): 282 doi: 10.1002/(ISSN)1097-461X
|
| [43] |
Scaranto J, Mallia G, Giorgianni S, et al. A quantum-mechanical study of the vinyl fluoride adsorbed on the rutile TiO2 (110) surface. Surf Sci, 2006, 600(2): 305 doi: 10.1016/j.susc.2005.10.032
|









Article views: 3559 Times PDF downloads: 29 Times Cited by: 0 Times
Received: 19 January 2016 Revised: 19 July 2016 Online: Published: 01 December 2016
| Citation: |
Xuechao Li, Jianhao Shi, Rundong Wan. The slabs for the rutile TiO2(110) surface[J]. Journal of Semiconductors, 2016, 37(12): 122003. doi: 10.1088/1674-4926/37/12/122003
****
X C Li, J H Shi, R D Wan. The slabs for the rutile TiO2(110) surface[J]. J. Semicond., 2016, 37(12): 122003. doi: 10.1088/1674-4926/37/12/122003.

|
| [1] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [2] |
Pang C L, Lindsay R, Thornton G. Chemical reactions on rutile TiO2 (110). Chem Soc Rev, 2008, 37(10): 2328 doi: 10.1039/b719085a
|
| [3] |
Harrison N M, Wang X G, Muscat J, et al. The influence of soft vibrational modes on our understanding of oxide surface structure. Faraday Discussions, 1999, 114: 305 doi: 10.1039/a906386b
|
| [4] |
Zhang Y, Lin W, Li Y, et al. A theoretical study on the electronic structures of TiO2: effect of Hartree-Fock exchange. J Phys Chem B, 2005, 109(41): 19270 doi: 10.1021/jp0523625
|
| [5] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theory Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [6] |
Charlton G, Howes P B, Nicklin C L, et al. Relaxation of TiO2 (110)-(1×1) using surface X-ray diffraction. Physl Rev Lett, 1997, 78(3): 495 doi: 10.1103/PhysRevLett.78.495
|
| [7] |
Hebenstreit E L D, Hebenstreit W, Diebold U. Structures of sulfur on TiO2 (110) determined by scanning tunneling microscopy, X-ray photoelectron spectroscopy and low-energy electron diffraction. Surf Sci, 2001, 470(3): 347 doi: 10.1016/S0039-6028(00)00849-9
|
| [8] |
Bennett R A, Pang C L, Perkins N, et al. Surface structures in the SMSI state; Pd on (1×2) reconstructed TiO2 (110). J Phys Chem B, 2002, 106(18): 4688 doi: 10.1021/jp0138328
|
| [9] |
Diebold U. Structure and properties of TiO2 surfaces: a brief review. Appl Phys A, 2003, 76(5): 681 doi: 10.1007/s00339-002-2004-5
|
| [10] |
Pillay D, Hwang G S. Structure of small Aun, Agn, and Cun clusters (n=2-4) on rutile TiO2 (110): a density functional theory study. Journal of Molecular Structure: THEOCHEM, 2006, 771(1): 129
|
| [11] |
Perron H, Domain C, Roques J, et al. Optimisation of accurate rutile TiO2 (110), (100), (101) and (001) surface models from periodic DFT calculations. Theoretical Chemistry Accounts, 2007, 117(4): 565 doi: 10.1007/s00214-006-0189-y
|
| [12] |
Busayaporn W. TiO2 (110) surface structure. University of Manchester, 2010
|
| [13] |
Lindsay R, Wander A, Ernst A, et al. Revisiting the surface structure of TiO2 (110): a quantitative low-energy electron diffraction study. Phys Rev Lett, 2005, 94(24): 246102 doi: 10.1103/PhysRevLett.94.246102
|
| [14] |
Cabailh G, Torrelles X, Lindsay R, et al. Geometric structure of TiO2 (110) (1×1): achieving experimental consensus. Phys Rev B, 2007, 75(24): 241403 doi: 10.1103/PhysRevB.75.241403
|
| [15] |
Busayaporn W, Torrelles X, Wander A, et al. Geometric structure of TiO2 (110) (1×1): confirming experimental conclusions. Phys Rev B, 2010, 81(15): 153404 doi: 10.1103/PhysRevB.81.153404
|
| [16] |
Ramamoorthy M, Vanderbilt D, King-Smith R D. First-principles calculations of the energetics of stoichiometric TiO2 surfaces. Phys Rev B, 1994, 49(23): 16721 doi: 10.1103/PhysRevB.49.16721
|
| [17] |
Bates S P, Kresse G, Gillan M J. A systematic study of the surface energetics and structure of TiO2 (110) by first-principles calculations. Surf Sci, 1997, 385(2): 386
|
| [18] |
Elliott S D, Bates S P. A first principles survey of stoichiometric (1×2) reconstructions on the rutile surface. Surf Sci, 2001, 495(3): 211 doi: 10.1016/S0039-6028(01)01328-0
|
| [19] |
Swamy V, Muscat J, Gale J D, et al. Simulation of low index rutile surfaces with a transferable variable-charge Ti-O interatomic potential and comparison with ab initio results. Surf Sci, 2002, 504: 115 doi: 10.1016/S0039-6028(01)01925-2
|
| [20] |
Vijay A, Mills G, Metiu H. Adsorption of gold on stoichiometric and reduced rutile TiO2 (110) surfaces. J Chem Phys, 2003, 118(14): 6536 doi: 10.1063/1.1557919
|
| [21] |
Wang Y, Hwang G S. Adsorption of Au atoms on stoichiometric and reduced TiO2 (110) rutile surfaces: a first principles study. Surf Sci, 2003, 542(1): 72
|
| [22] |
Sano H, Mizutani G, Wolf W, et al. Ab initio calculation of optical second harmonic generation at the rutile TiO2 (110) surface. Phys Rev B, 2004, 70(12): 125411 doi: 10.1103/PhysRevB.70.125411
|
| [23] |
Thompson S J, Lewis S P. Revisiting the (110) surface structure of TiO2: a theoretical analysis. Phys Rev B, 2006, 73(7): 073403 doi: 10.1103/PhysRevB.73.073403
|
| [24] |
Hameeuw K J, Cantele G, Ninno D, et al. The rutile TiO2 (110) surface: Obtaining converged structural properties from first-principles calculations. J Chem Phys, 2006, 124(2): 024708 doi: 10.1063/1.2136158
|
| [25] |
Labat F, Baranek P, Adamo C. Structural and electronic properties of selected rutile and anatase TiO2 surfaces: an ab initio investigation. J Chem Theor Comput, 2008, 4(2): 341 doi: 10.1021/ct700221w
|
| [26] |
Thompson S J, Lewis S P. Deconstructing the structural convergence of the (110) surface of rutile TiO2. In: Computer simulation studies in condensed-matter physics XIX. Springer Berlin Heidelberg, 2009, 123: 26
|
| [27] |
Slater J C. The self-consistent field for molecules and solids. New York: McGraw-Hill, 1974
|
| [28] |
Vosko S H, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Canadian J Phys, 1980, 58(8): 1200 doi: 10.1139/p80-159
|
| [29] |
Binkley J S, Pople J A, Hehre W J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J Amer Chem Soc, 1980, 102(3): 939 doi: 10.1021/ja00523a008
|
| [30] |
Dobbs K D, Hehre W J. Molecular orbital theory of the properties of inorganic and organometallic compounds 5. Extended basis sets for first-row transition metals. J Comput Chem, 1987, 8(6): 861 doi: 10.1002/(ISSN)1096-987X
|
| [31] |
Mattsson A, Hu S, Hermansson K, et al. Adsorption of formic acid on rutile TiO2 (110) revisited: an infrared reflection-absorption spectroscopy and density functional theory study. J Chem Phys, 2014, 140(3): 034705 doi: 10.1063/1.4855176
|
| [32] |
Chang T Y, Tanaka Y, Ishikawa R, et al. Direct imaging of Pt single atoms adsorbed on TiO2 (110) surfaces. Nano Lett, 2013, 14(1): 134
|
| [33] |
Rangan S, Ruggieri C, Bartynski R, et al. Densely packed ZnTPPs monolayer on the rutile TiO2 (110)-(1×1) surface: adsorption behavior and energy level alignment. J Phys Chem C, 2016, 120(8): 4430 doi: 10.1021/acs.jpcc.5b12736
|
| [34] |
Ong S V, Khanna S N. Theoretical studies of the stability and oxidation of Pdn (n=1-7) clusters on rutile TiO2 (110): adsorption on the stoichiometric surface. J Phys Chem C, 2012, 116(4): 3105 doi: 10.1021/jp212504x
|
| [35] |
Zhang Z, Tang M, Wang Z T, et al. Imaging of formaldehyde adsorption and diffusion on TiO2 (110). Topics in Catalysis, 2015, 58(2/3): 103
|
| [36] |
García-Mota M, Vojvodic A, Abild-Pedersen F, et al. Electronic origin of the surface reactivity of transition-metal-doped TiO2 (110). J Phys Chem C, 2012, 117(1): 460
|
| [37] |
Sasahara A, Tomitori M. XPS and STM study of Nb-doped TiO2 (110)-(1×1) surfaces. J Phys Chem C, 2013, 117(34): 17680 doi: 10.1021/jp4057576
|
| [38] |
Frisch M, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision C. 01, Gaussian. Inc., Wallingford, CT, 2009
|
| [39] |
Materials Studio version 4.4 from Accelrys http://accelrys.com/events/webinars/materials-studio-44/
|
| [40] |
Heyd J, Scuseria G E. Efficient hybrid density functional calculations in solids: assessment of the Heyd-Scuseria-Ernzerh of screened Coulomb hybrid functional. J Chem Phys, 2004, 121(3): 1187 doi: 10.1063/1.1760074
|
| [41] |
Bredow T, Giordano L, Cinquini F, et al. Electronic properties of rutile TiO2 ultrathin films: odd-even oscillations with the number of layers. Phys Rev B, 2004, 70(3): 035419 doi: 10.1103/PhysRevB.70.035419
|
| [42] |
Evarestov R A, Bandura A V. Hartree-Fock calculations of electronic structure of (110)-surface of rutile TiO2: comparison of single (2D) and periodic (3D) slab models. International Journal of Quantum Chemistry, 2004, 96(3): 282 doi: 10.1002/(ISSN)1097-461X
|
| [43] |
Scaranto J, Mallia G, Giorgianni S, et al. A quantum-mechanical study of the vinyl fluoride adsorbed on the rutile TiO2 (110) surface. Surf Sci, 2006, 600(2): 305 doi: 10.1016/j.susc.2005.10.032
|

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2

 DownLoad:
DownLoad:

