


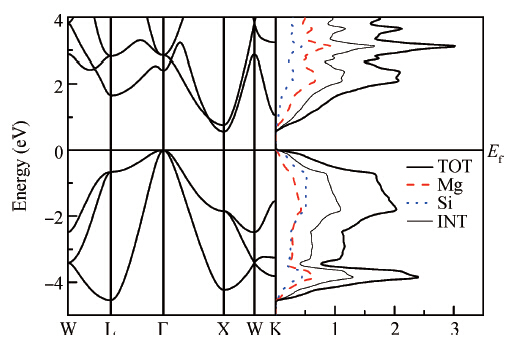
Fig. 1.
Calculated energy bands and density of states (DOS, in state/eV per unit cell) of {Mg$_2$Si} within mBJ.
SEMICONDUCTOR MATERIALS
Corresponding author: Sandong Guo , Email: guosd@cumt.edu.cn
Abstract: The electronic structures of Mg2X (X = Si, Ge, Sn) have been calculated by using generalized gradient approximation, various screened hybrid functionals, as well as Tran and Blaha's modified Becke and Johnson exchange potential. It was found that the Tran and Blaha's modified Becke and Johnson exchange potential provides a more realistic description of the electronic structures and the optical properties of Mg2X (X = Si, Ge, Sn) than else exchange-correlation potential, and the theoretical gaps and dielectric functions of Mg2X (X = Si, Ge, Sn) are quite compatible with the experimental data. The elastic properties of Mg2X (X = Si, Ge, Sn) have also been studied in detail with the generalized gradient approximation, including bulk modulus, shear modulus, Young's modulus, Poisson's ratio, sound velocities, and Debye temperature. The phonon dispersions of Mg2X (X = Si, Ge, Sn) have been calculated within the generalized gradient approximation, suggesting no structural instability, and the measurable phonon heat capacity as a function of the temperature has been also calculated.
Keywords: semiconductor, optical properties, elastic properties, phonon
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] |
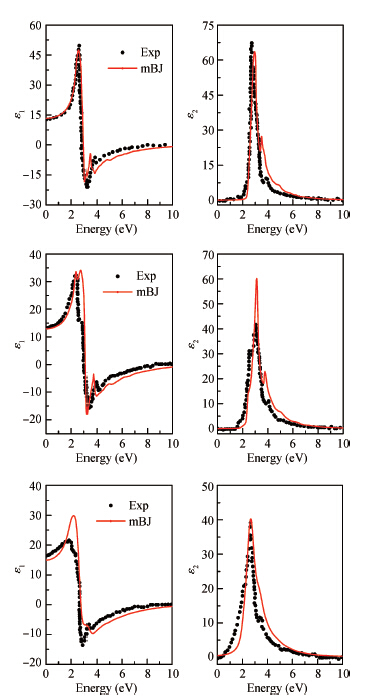

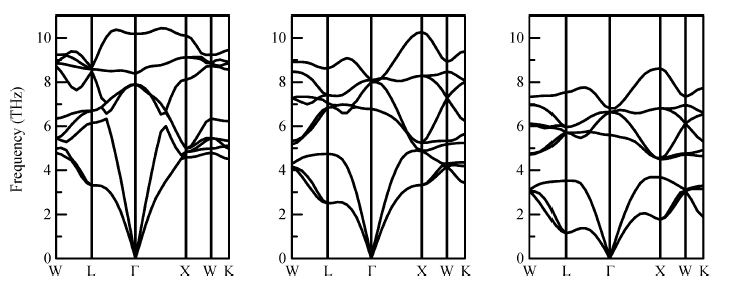
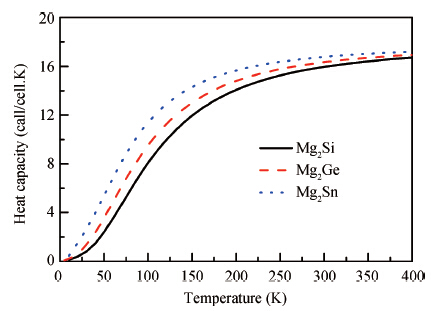
Table 1. Calculated energy gaps (using GGA, sX-LDA, PBE0, HSE03, HSE06 and mBJ) of Mg$_2$X (X $=$ Si, Ge, Sn), corresponding experimental results and other calculated values.
 DownLoad: CSV
DownLoad: CSV
Table 2. Elastic constants $C_{ij}$ (in GPa), bulk modulus $B$ (in GPa) compared to experimental values[39, 40] given in parentheses and anisotropy factor $A$.
DownLoad: CSV
Table 3. The calculated shear modulus $G$ (in GPa), Young's modulus $E$ (in GPa), Poisson's ratio $\nu$, $B/G$.
DownLoad: CSV
Table 4. The calculated sound velocities $v_{\rm l}$, $v_{\rm t}$ and $v_{\rm m}$ (in m/s), $\rho$ (in kg/m$^3$) and Debye temperature $\theta_{\rm D}$ (in K).
DownLoad: CSV
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] |









Article views: 3267 Times PDF downloads: 23 Times Cited by: 0 Times
Received: 03 November 2014 Revised: Online: Published: 01 May 2015
| Citation: |
Sandong Guo . First-principles calculations of Mg2X (X = Si, Ge, Sn) semiconductors with the calcium fluorite structure[J]. Journal of Semiconductors, 2015, 36(5): 053002. doi: 10.1088/1674-4926/36/5/053002
****
S D Guo. First-principles calculations of Mg2X (X = Si, Ge, Sn) semiconductors with the calcium fluorite structure[J]. J. Semicond., 2015, 36(5): 053002. doi: 10.1088/1674-4926/36/5/053002.

|
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] |

 WeChat ID
WeChat ID
Journal of Semiconductors © 2017 All Rights Reserved 京ICP备05085259号-2






